DNA甲基化芯片分析有不少R包实现,如:minfi、lumi以及ChAMP等,我只粗略看过minfi和ChAMP,发现ChAMP的功能更加齐全以及使用也较为简单,并且其也集成了minfi包的部分功能,所以下面以ChAMP包作为学习对象 #### ChAMP包的安装
source("https://bioconductor.org/biocLite.R")
options(BioC_mirror="http://mirrors.ustc.edu.cn/bioc/")
biocLite("ChAMP")如果是墙内,最好加上options选择中科大镜像,因为有好多依赖包需要安装
ChAMP包也依赖了minfi包的一些功能,还似乎安装了好多配套注释文件的包,省的以后需要时再去找了,后续需要的话直接加载即可了
读入数据
这里有个问题,由于ChAMP包以及minfi包读入文件时,除了需要.idat文件外,还需要sample_sheet.csv文件,里面记录了样本信息、样本分组信息、每个GSM文件对应哪个样本等信息。但是之前给的那个测试数据我在GEO上下载的RAW文件里没有该样本信息文件。后来也尝试了选择其他GSE号的数据,也未发现有该文件,最终暂时放弃尝试了,决定选择ChAMP自带的450K芯片的测试数据(理论上illumina下机后除了有.idat文件外,还有sample_sheet.csv文件的)
先下载测试数据
testDir=system.file("extdata",package="ChAMPdata")
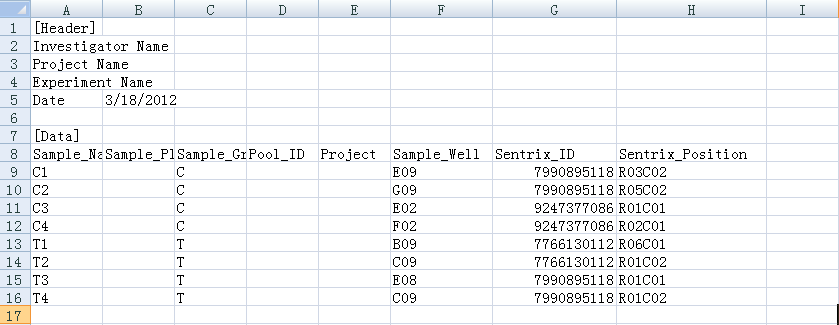
myLoad <- champ.load(testDir,arraytype="450K")数据可以在R包library/ChAMPdata/extdata文件夹下找到,里面除了.idat文件外,还有一个lung_test_set.csv样本信息文件;所以我猜想,尽管有些GSE数据没有样本信息.csv文件,我们可以根据lung_test_set.csv格式模仿写一个(Header部分可有可无,但Data部分几个关键的列必须要有),前提当然要知道样本分组信息(如果不知道的话,就编一个吧。。。),模板如下图所示:
除了上述文件外,还有一个文件比较重要的是450K注释文件,如果是GEO下载的数据化,就是XXXHumanMethylation450XXX.csv文件,里面有详细的每个探针的数据,比如探针的ID、不同探针Type的序列,在染色体上的定位信息以及注释信息等
数据过滤
准备好上述文件后,接下来则是读入数据,使用ChAMP包的champ.load函数,按照其说法,champ.load函数其实做了两个步骤,第一是读入数据(champ.import函数),第二是过滤数据(champ.filter函数),所以我们需要了解champ.load以什么标准过滤: >1. 过滤掉detection p-value大于0.01的探针 >2. 过滤掉在至少5%样本中bead count小于3的探针 >3. 过滤掉非GpC位点的探针 >4. 过滤掉所有SNP相关的探针 >5. 过滤掉multi-hit探针,即映射到多个位置的 >6. 过滤掉X和Y染色体上的探针
以上标准均是champ.load的默认参数,如果是850K芯片还需加上arraytype="850K",因为默认是450K芯片。。
myLoad <- champ.load("E:/R-3.4.1/library/ChAMPdata/extdata")读入数据时会出现很多信息,其中可以看到有613个control probes,这个探针主要用于计算芯片数据的可信度Detect P value,具体解释可参考DNA Methylation中的Detect P value解析
Fetching NEGATIVE ControlProbe. Totally, there are 613 control probes in Annotation. Your data set contains 613 control probes.
还有不少过滤过程的信息,基本上是根据你设定的过滤参数来的
做完上述步骤,ChAMP包还提供了CpG.GUI()函数用于展示distribution of CpGs on chromosome, CpG island, TSS reagions. e.g,其实是几张简单展示myLoad$beta分布的图片,但是界面很友好,也难怪依赖包里要shiny包了
CpG.GUI(CpG = rownames(myLoad$beta))对于
数据质控QC
ChAMP包不愧是集成度很高的DNA甲基化分析的R包,啥功能都有。。champ.QC()和QC.GUI函数用于检查看看过滤后的数据是否符合要求,能否进行下一步的分析。
champ.QC()函数结果三张图,如有mdsPlot (Multidimensional Scaling Plot),主要看看不同分组样本是否分开;densityPlot,每个样本的beta值的分布图,主要看看有无异常的样本;dendrogram,样本的聚类图
champ.QC(beta = myLoad$beta, pheno = myLoad$pd$Sample_Name)上述代码我的电脑会报错:LAPACK routines cannot be loaded,网上查了下,似乎是电脑的原因,具体图片样式可看R包说明文档,主要也是根据myLoad\(beta数据以及myLoad\)pd$Sample_Name样本信息来绘制图片的
QC.GUI(beta=myLoad$beta)这个也是报同样的错误,但ChAMP包的结果可视化做的确实蛮好的
数据标准化
由于Illumina beadarrays是由两套探针,使用不同的杂交方法,所以如果上述QC结果中发现type-I探针和type-II探针有较大偏差,则需进行校正;简单的说就是消除测序芯片上的两种测序技术之间的差异。ChAMP包标准化方法有BMIQ、SWAN、PBC以及FunctionalNormliazation,我这里就先选择默认的BMIQ,后续如果有需求再看看其他标准化方法
myNorm <- champ.norm(beta=myLoad$beta,arraytype="450K",cores=5)SVD plot
SVD(singular value decomposition) 只为检测变异组分的显著性,主要用于查看变异的主要成分是生物学处理等影响的,还是技术因素所造成的,如果是后者则需要进行后续的批次校正。从下面代码也可看出其主要根据myNorm(校正后的探针的beta)和pd(样本信息)来计算变异程度
champ.SVD(beta=myNorm,pd=myLoad$pd)批次效应校正
如果上面SVD Plot显示红色块并不在Sample_Group行,而是较多的在Array行,那么则需要对这次数据进行批次校正,使用champ.runCombat函数,batchname则是根据上述SVD plot出现的红块位置而定,决定哪个batch factor需要被校正;这个过程比较耗时,做完后最好再用champ.SVD检查下
myCombat <- champ.runCombat(beta=myNorm,pd=myLoad$pd,batchname=c("Slide"))甲基化探针差异分析
比较常见的差异甲基化分析是DMP(Differential Methylation Probe),用于找出差异的甲基化位点,然后ChAMP包已经将分析过程(主要还是基于Limma包,这个包是专门用于分析芯片差异表达的包);然后根据你的分组信息,获得差异甲基化位点;最后用DMP.GUI查看下结果
myDMP <- champ.DMP(beta = myNorm,pheno=myLoad$pd$Sample_Group)
DMP.GUI(DMP=myDMP[[1]],beta=myNorm,pheno=myLoad$pd$Sample_Group)除了可以分析差异甲基化位点外,还可以分析DMR(Differential Methylation Regions),用于找出差异甲基化片段?用的函数也很好记champ.DMR(),该函数现在只支持两组样本进行差异分析
myDMR <- champ.DMR(beta=myNorm,pheno=myLoad$pd$Sample_Group,method="Bumphunter")这里需要注意的是method的选择,主要有Bumphunter, ProbeLasso and DMRcate三者可以选择,区分的话 >1. Bumphunter,groups all probes into small clusters (or regions), then applies random permutation method to estimate candidate DMRs,不用依赖上述差异位点分析的结果 >2. ProbeLasso,the final data frame is distilled from a limma output of probes and their association statistics,必须要有champ.DMP()的结果作为输入 >3. DMRcate,a new method recently incoporated into ChAMP,pass the square of the moderated t statistic calculated on each 450K probe to DMR-finding function(这个还是看文档吧)
除了DMR外还有一个DMB(Differential Methylation Blocks),也是用来识别大片段的差异甲基化
myBlock <- champ.Block(beta=myNorm,pheno=myLoad$pd$Sample_Group,arraytype="450K")其他分析类型
除了上述比较常规差异甲基化分析的外,还有GSEA富集分析(Gene Set Enrichment Analysis)、网络分析(Differential Methylated Interaction Hotspots)和拷贝数变异(Copy Number Variation),ChAMP包都集成到一个个函数里了,用起来确实比较方便
Summary
本来这个笔记应该是实际数据操作的过程,自从数据质控QC开始报错后,后面只要用到LAPACK的函数均报错了,找了好久都没找到在windows下的解决办法(只能这过段时间在服务器上跑跑看了)。所以这笔记变成了ChAMP包文档笔记了,顺着文档粗略的扫了一遍,没细读,以后需要的时候再补了
最后上述的BUG还是解决了,原来报错的是因为LAPACK routines没有被加载,但是真正的原因则是因为 maximal number of DLLs reached,出现这个的原因是ChAMP包加载很多DLLs,而R默认的最大DLLs是100,如果超过100个了,那么就不让继续加载新的DLL。
因此解决办法是更改R的默认设置,让R_MAX_NUM_DLLS变量的值设为100+,之前查的解决办法都是在linux或者mac上的,今天终于让我找到在windows下的一种解决办法,就是在设置电脑环境变量中新增一个变量R_MAX_NUM_DLLS,并设置值为200,然后重新启动R,然后上述的那些ChAMP包都能正常工作了。可以好好看看整个流程的结果了
参考资料:
The Chip Analysis Methylation Pipeline
计算表观学公众号:Illumina 850K(EPIC)甲基化芯片及其分析工具ChAMP
生信技能树公众号:850K甲基化芯片数据的分析
Shiny和Plotly实现可交互DNA甲基化分析包ChAMP
DNA Methylation中的Detect P value解析
读取DNA甲基化IDAT文件
集识别差异甲基化和拷贝数变异于一体的R包——ChAMP
bioconductor的minfi处理甲基化数据
Illumina HumanMethylation450 BeadChip (甲基化450k芯片) 预处理初探
450K甲基化芯片数据处理传送门
本文出自于http://www.bioinfo-scrounger.com转载请注明出处