KeepNotes blogStay hungry, Stay Foolish.2024-09-19T02:08:37.496Zhttp://www.bioinfo-scrounger.com/KaiHexoMiettinen-Nurminen (MN) Test in R and SAShttp://www.bioinfo-scrounger.com/archives/mn_test/2024-09-19T02:03:21.000Z2024-09-19T02:08:37.496ZMiettinen-Nurminen (MN) method has increasingly been requested by regulatory agencies, rather than the traditional Wald method that is based on the asymptotic normal distribution. This is particularly relevant for non-inferiority trials where it's appropriate for the variance to be estimated under the null hypothesis. Calculate MN Test Statistics within PROC FREQ.
As can be seen from the above description, if we would like to get the CI of M&N method, we should compute the root of the maximum likelihood estimations with the closed-form solution provided by M&N. Referring to the following method from metalite.ae rate-compare vignettes, we can create an equation p2 - p1 = delta and the chi-square statistic to find out the root.
MN_formula2
As can be seen from the above explanation, if we would like to get the confidence interval (CI) of M&N method, we should compute the root of the maximum likelihood estimations with the closed-form solution provided by M&N. Referring to the following method, we can construct an equation to find out the root from the chi-square distribution with 1 degree of freedom. So I roughly understand that the delta can be regarded as the CI when the equation of p1 - p2 - delta divided by variance is equivalent to the chi-square statistic. Therefore, I create a function so that I can obtain the root using a solving process.
mn_root <- function(delta, alpha = 0.05) { diff <- p1 - p2 - delta theta <- n2 / n1 a <- 1 + theta b <- -(1 + theta + p1 + theta * p2 + delta * (theta + 2)) c <- delta^2 + delta * (2 * p1 + theta + 1) + p1 + theta * p2 d <- -p1 * delta * (1 + delta) v <- b^3 / (27 * a^3) - b * c / (6 * a^2) + d / (a * 2) # here should be '-' instead of '+' inside the square root. u <- ifelse(v > 0, 1, -1) * sqrt(b^2 / (9 * a^2) - c / (a * 3)) w <- (pi + acos(v / u^3)) / 3 p1d <- 2 * u * cos(w) - b / (a * 3) p2d <- p1d - delta # n <- n1 + n2 var <- (p1d * (1 - p1d) / n1 + p2d * (1 - p2d) / n2) * (n1 + n2) / (n1 + n2 - 1) chisq <- diff^2 / var return(chisq - qchisq(1 - alpha, 1))}
Now I also create an example data as shown below to compute the risk difference (RD) of response rate between two treatments.
By this example data, I can know the sample size and response rate for each treatment group. And then in order to obtain the root, someone will use the bisection method but I feel that we can just use the uniroot() to solve the single root, or rootSolve::uniroot.all() to get multiple roots.
Then I use the metalite.ae::rate_compare() to check the result from the above self-defined function. Both of the upper and lower values of CI are consistent.
metalite.ae::rate_compare(response ~ treatment, data = ana)## est z_score p lower upper## 1 0.4 5.759051 4.229411e-09 0.269662 0.5165743
According to the stratified M&N test, I will just talk about it briefly here. If you would like to use the Miettinen-Nurminen stratified method as described in the 1985 paper by SAS, unfortunately the current SAS version cannot support it yet. Although I can use COMMONRISKDIFF(CL=score) to compute the CI of risk difference, the results are not the MN 1985 method that is referred to as Agresti Score at the summary level. More discussions can be found in Confidence limits using the Miettinen-Nurminen stratified method as described in the 1985 paper, and Calculate MN Test Statistics within PROC FREQ.
The basic SAS and R codes for example data used to compute the unstratified and stratified Miettinen and Nurminen test are provided below for reference. It should be emphasized that the results of stratified M&N results are not consistent.
R code:
# Unstratified M&N testmetalite.ae::rate_compare( formula = response ~ treatment, data = ana)# Stratified M&N testmetalite.ae::rate_compare( formula = response ~ treatment, data = ana, strata = stratum, weight = "ss")## est z_score p lower upper## 1 0.3998397 5.712797 5.556727e-09 0.2684383 0.5172779
MN_formula_SAS]]>
<p>Miettinen-Nurminen (MN) method has increasingly been requested by regulatory agencies, rather than the traditional Wald method that is based on the asymptotic normal distribution. This is particularly relevant for non-inferiority trials where it's appropriate for the variance to be estimated under the null hypothesis. <a href="https://communities.sas.com/t5/SAS-Product-Suggestions/Calculate-MN-Test-Statistics-within-PROC-FREQ/idi-p/933198" target="_blank" rel="noopener">Calculate MN Test Statistics within PROC FREQ</a>.</p>
Learning Bisection Methodhttp://www.bioinfo-scrounger.com/archives/bisection/2024-07-03T12:09:23.000Z2024-07-03T12:14:17.442ZWhen I learned the Miettinen and Nurminen Test algorithm from the rate-compare article (Unstratified and Stratified Miettinen and Nurminen Test), I found that its CI is given by the roots of an equation. In order to reproduce its algorithm, I'd like to learn more about the Bisection method first.
The bisection method is an approach to finding the root of a continuous function on an interval. Its principle can be shown below (referring from https://rpubs.com/aaronsc32/bisection-method-r).
The method takes advantage of a corollary of the intermediate value theorem called Bolzano's theorem which states that if the values of f(a) and f(b) have opposite signs, the interval must contain at least one root. The iteration steps of the bisection method are relatively straightforward, however; convergence towards a solution is slow compared to other root-finding methods.
Thus the algorithm can be divided into the following steps. - Calculate the minpoint m=(a+b)/2 based on the interval with the lower bound a and higher bound b. - Get the value of function f at midpoint. If f(a) and f(m) have opposite signs, then b point is replaced by the computed midpoint, or if f(b) and f(m) have opposite signs, a is replaced by midpoint. This step ensures the root of function can be kept within the new interval. - Iterate over the above steps 1-2 until the difference between a and b is small enough or the defined iterating number is reached, at which midpoint m is the root.
According to the above steps, we can write a function to implement the bisection method in R. Assume we have the following function f to be solved.
Then we create a function with bisection algorithm and find the root.
bisect_func <- function(f, a, b, tol = 1e-5, n = 100) { i <- 1 while (i <= n) { m <- (a + b) / 2 if (abs(b - a) < tol | f(m) == 0) { break } if (f(a) * f(m) <0) { b <- m } else if (f(b) * f(m) < 0) { a <- m } else { stop("f(a) and f(b) must have opposite sign.") } i <- i + 1 if (i > n) { warning("The max iterations reached, maybe the lower and upper bound should be changed and try again.") } } root <- (a + b) / 2 return(root)}bisect_func(f, 0, 2, tol = 0.001)[1] 0.6293945
And the same outcome can be obtained from the mature function cmna::bisection().
cmna::bisection(f, 0, 2)[1] 0.6293945
Actually this f function should have two roots, but the bisect_func() or cmna::bisection() functions trend to have the root on the lower bound side. In this case, we must either run the bisection function multiple times for different intervals, or use another function able to finding multiple roots.
# two roots in (0,1) and (1,2)bisect_func(f, 0, 1, tol = 0.001)[1] 0.6293945bisect_func(f, 1, 2, tol = 0.001)[1] 1.447754# using uniroot.all() functionrootSolve::uniroot.all(f, c(0, 2), n = 100)[1] 0.6293342 1.4480258
Above is my brief summary and hope it can be useful. And in the following post I will learn how to use bisection method or uniroot function to solve in the Miettinen-Nurminen (Score) Confidence Limits.
]]>
<p>When I learned the Miettinen and Nurminen Test algorithm from the <code>rate-compare</code> article (<a href="https://cran.r-project.org/web/packages/metalite.ae/vignettes/rate-compare.html" target="_blank" rel="noopener">Unstratified and Stratified Miettinen and Nurminen Test</a>), I found that its CI is given by the roots of an equation. In order to reproduce its algorithm, I'd like to learn more about the Bisection method first.</p>
Shift Value for MCMC imputation in Tipping Point Analysishttp://www.bioinfo-scrounger.com/archives/mcmc_tpa/2024-07-03T12:05:18.000Z2024-07-03T12:14:18.306ZThis is a continuation of the previous article, Tipping Point Analysis in Multiple Imputation Using SAS. In the last post, we talked about the tipping point analysis in monotone imputation, but how to implement TPA in MCMC imputation since the MNAR statement can only support the shift adjustment in monotone and FCS.
Before that, let's have a look at the process of imputation and shift adjustment in the monotone method. By default, the missing values are imputed sequentially in the order specified in the VAR statement. For example, the following MI procedure uses the regression method to impute each variable by its previous variables, which means variable week8 is imputed by effects from basval to week6, and variable week6 is imputed by effects from basval to week4. The variable basval is not imputed since it is the leading variable in the VAR statement. And you can also specify difference imputation methods in monotone statement for each variable, more details can be found in the SAS documentation
proc mi data=low1_wide seed=12306 nimpute=10 out=imp_mnar2; class trt; by trt; var basval week1 week2 week4 week6 week8; monotone reg; mnar adjust (week8 / shift=-1 adjustobs=(trt='1')); mnar adjust (week8 / shift=1 adjustobs=(trt='2'));run;
According to above logic, I feel we can manually add shift values instead of MNAR statement in the MCMC imputation by following the steps.
Assuming the missingness pattern is arbitary rather than monotone, the MCMC full-data imputation approach is selected.
Get the imputed datasets and find out where the missing data is in the original dataset, such as the location of the subject and visit.
Adjust the week8 variable with the shift values, for each treatment (if needed).
I have tested the logic by comparing the results of the MNAR statement against manual adjusting. That is identical, so use the SAS code as shown below for MCMC imputation.
proc mi data=low1_wide seed=12306 nimpute=10 out=imp_mcmc; mcmc impute=full niter=1000 nbiter=1000; by trt; var basval week1 week2 week4 week6 week8;run;proc sort; by patient _imputation_; run;proc sort data=low1_wide out=low1_wide_w8(keep=patient week8 rename=(week8=w8)); by patient;run;data imp_mcmc_shift; merge imp_mcmc low1_wide_w8; by patient; if missing(w8) then do; if trt='1' then week8=week8-1; else if trt='2' then week8=week8+1; end; drop w8;run;
]]>
<p>This is a continuation of the previous article, <a href="https://www.bioinfo-scrounger.com/archives/tpa_mi/">Tipping Point Analysis in Multiple Imputation Using SAS</a>. In the last post, we talked about the tipping point analysis in monotone imputation, but how to implement TPA in MCMC imputation since the <code>MNAR</code> statement can only support the shift adjustment in monotone and FCS.</p>
Tipping Point Analysis in Multiple Imputation using SAShttp://www.bioinfo-scrounger.com/archives/tpa_mi/2024-07-01T12:37:07.000Z2024-07-01T12:38:53.462ZThe tipping point analysis has been a useful sensitivity analysis for multiple imputation to assess the robustness of the deviations from the MCAR or MAR assumptions. It aims to find out how severe departures from MAR will overturn the conclusions from primary analysis. If the departures are considered unlikely, this can give strong evidence supporting the treatment effect found in the primary analysis under the MAR assumptions.
In some ways, tipping point analysis can be regarded as supplementary to multiple imputation, allowing us to explore the outcomes from different scenarios by applying the shift parameters, such as decreasing the treatment effect while increasing the control group effect. The end goal is to identify the point at which the primary analysis result with multiple imputation becomes non-significant.
The tipping point analysis can be easily implemented using SAS procedure PROC MI and the brief instructions can be found in Sensitivity Analysis with the Tipping-Point Approach where it uses the MONOTONE statement to impute the missing data and the MNAR statement to adjust the imputed data by a range of specified shift parameters. Thus the analysis steps can be described as follows:
If the missing pattern you detect is intermittent rather than monotone, you should fill in the missing data using MCMC statement so that the pattern can be transformed into monotone.
If the missing data has monotone pattern, impute it using MONOTONE or FCS method in Proc MI under the MAR assumptions. And specify the shift parameters in MNAR statement to adjust the imputed value for observations in any treatment group as needed.
Based on the imputed datasets from step 2, apply the pre-specified models to analyze each dataset and obtain the statistical results.
Combine all of the results from step 3 by Rubin's rule using Proc MIANALYZE and make statistical inferences.
Repeat steps 2-4 by adjusting the shift parameters to get a set of inference outcomes to identify the tipping point that overturns the conclusions from significant to non-significant.
Here, let's look at the SAS code below. I will use the example missing dataset (low1.sas7bdat) from Mallinckrodt et al. (https://journals.sagepub.com/doi/pdf/10.1177/2168479013501310). And transform it to fit the follow-up analysis requirement.
proc sort data=low1; by patient trt basval; run;proc transpose data=low1 out=low1_wide(drop=_name_) prefix=week; by patient trt basval; id week; var change;run;
As I have checked this dataset has a monotone missing pattern, so I don't need to go through step 1. But if you need or want to achieve that, try the code below. Please note that we have generated nimpute=10 imputed datasets in this step, we just need to apply nimpute=1 in the next monotone imputation and MNAR adjust steps.
/* Step 1: Achieve Monotone Missing Data Pattern */proc sort data=low1_wide; by trt; run;proc mi data=low1_wide seed=12306 nimpute=10 out=imp_mono; mcmc impute=monotone nbiter=1000 niter=1000; by trt; var basval week1 week2 week4 week6 week8;run;/* Step2: MAR imputation and MNAR adjustation at week 8 visit for each group. */proc sort data=imp_mono; by _imputation_ trt; run;proc mi data=imp_mono seed=12306 nimpute=10 out=imp_mnar2; class trt; by _imputation_ trt; var trt basval week1 week2 week4 week6 week8; monotone reg; mnar adjust (week8 / shift=-1 adjustobs=(trt='1')); mnar adjust (week8 / shift=1 adjustobs=(trt='2'));run;
Back to assuming the example dataset has a monotone pattern, we just jump into step 2. Here, I suppose the primary endpoint is the change from baseline at week 8. So the prior visits are imputed under the MAR assumption using MONOTONE REG, and the week 8 visit will have an additional MNAR process where the trt=1 group is made better by adding a delta (shift=-1) while the trt=2 group is made worse by a delta (shift=1) as the lower value implies the better treatment effect. I also want to impute the datasets separately for each treatment group, so I set the BY statement to trt.
/* Step2: MAR imputation using MONOTONE */proc sort data=low1_wide; by trt; run;proc mi data=low1_wide seed=12306 nimpute=10 out=imp_mnar2; class trt; by trt; var basval week1 week2 week4 week6 week8; monotone reg; mnar adjust (week8 / shift=-1 adjustobs=(trt='1')); mnar adjust (week8 / shift=1 adjustobs=(trt='2'));run;
From the SAS documentation, the MNAR statement is applicable only if it is used along with the MONOTONE and FCS statement. So why did I choose the former one here instead of the latter? Refer to this article (Application of Tipping Point Analysis in Clinical Trials using the Multiple Imputation Procedure in SAS), it states that only MONOTONE can provide us with the exact shift value we specified in imputed values straightforwardly, whereas the FCS needs a bit trick processing although it has more advantages somewhere. P.S. I did check it, indeed as mentioned above.
Now we generate 10 imputed datasets with a single shift value, and then these complete datasets are analyzed using the ANCOVA model, and the results are combined using Proc MIANALYZE, which is a typical multiple imputation process. So we don't need to describe them as details, just put all of above steps into one macro so that we can get a set of results using different shift values.
%macro mi_tpa(ind=, smin=, smax=, sinc=, out=); /* Create a set of shift values */ %let ncase=%sysevalf((&smax. - &smin.) / &sinc., ceil); data &out.; set _null_; run; /* Looping implement each shift for monotone imputation */ %do i=0 %to &ncase.; %let k=%sysevalf(&smin. + &i. * &sinc.); proc sort data=&ind.; by trt; run; proc mi data=&ind. seed=12306 nimpute=10 out=imp_mnar; class trt; by trt; var basval week1 week2 week4 week6 week8; monotone reg; mnar adjust (week8 / shift=-&k. adjustobs=(trt='1')); mnar adjust (week8 / shift=&k. adjustobs=(trt='2')); run; proc sort; by _imputation_; run; /* Step 3: Implement ANCOVA model for each imputation*/ ods output lsmeans=lsm diffs=diff; proc mixed data=imp_mnar; by _imputation_; class trt(ref='1'); model week8=basval trt /ddfm=kr; lsmeans trt / cl pdiff diff; run; /* Step 4: Pooling model results */ ods output ParameterEstimates=combined_diff; proc mianalyze data=diff; by trt _trt; modeleffects estimate; stderr stderr; run; /* Output results */ data mnar; set combined_diff; shift=&k.; run; data &out.; set &out. mnar; run; %end;%mend;
Here, we assume that the tipping point that reverses the conclusion is between 0 and 5. Thus I define the range from 0 to 5 with an interval of 0.5. The following code performs the MNAR adjustment with each of the shift values, like 0, 0.5, 1,...,4.5, 5.
Regarding each shift value, the smallest value where the p-value is no longer significant is identified as the tipping point (in the red box). The next question is whether the shift value is reasonable in clinical practice. If that is not reasonable or unlikely, it can provide strong support for out primary conclusion.
]]>
<p>The tipping point analysis has been a useful sensitivity analysis for multiple imputation to assess the robustness of the deviations from the MCAR or MAR assumptions. It aims to find out how severe departures from MAR will overturn the conclusions from primary analysis. If the departures are considered unlikely, this can give strong evidence supporting the treatment effect found in the primary analysis under the MAR assumptions.</p>
Simply Understanding Log Rank Testhttp://www.bioinfo-scrounger.com/archives/logrank-test/2024-06-26T15:23:23.000Z2024-06-26T15:38:49.216ZThe logrank test is the most commonly used statistical test in clinical trials to compare the survival distributions in different treatment groups. We usually just use the logrank results to test whether there is a difference between two survival curves. But what does this difference mean?
First of all, the difference is a statistical concept, so we need a statistical hypothesis test to get the p-value in order to determine whether the difference is significant.
Null hypothesis: The two treatment groups have identical survival distributions.
Alternative hypothesis: The two treatment groups have different survival distributions.
Now the question turns to how to get the p-value. Before that we should obtain a test statistic that follows the a chi-squared distribution. Thus the question can be simplified to how to compute the test statistic, with details and equations available in the references above. Let's create an example data used to display the whole calculation process in R.
Suppose we have group 1 and group 2 including six time to event data. The table shows the survival time in t columns and the evt column tells us if an event occurred (evt=1) or the case censored (evt=0) in the corresponding time.
Then we need to convert above datasets to unique survival times, summarize the number of events (m column) and consors (q column), and add one column to represent the risk number (n column) at each time. Then outer join two dataset using t variable and filter the invalid time as the time without events cannot offer any meaningful information for test statistic.
To get the statistic, we should firstly compute the so-called expected value (e1 or e2), the difference (me1 or me2) of the observed value (m1 or m2) minus the expected values, and the variance (v).
Base on above computations, now we can simply calculate the test statistic, that is 1.6205 in our example. Then the p-value can be determined using the chi-squared distribution with one degree of freedom (number of groups minus 1).
Now that we should have a basic knowledge of logrank, and let us check with those found in the mature R package survival.
data <- bind_rows( data.frame( t = c(3.1, 6.8, 9, 9, 11.3, 16.2), m = c(1, 0, 1, 1, 0, 1) ), data.frame( t = c(8.7, 9, 10.1, 12.1, 18.7, 23.1), m = c(1, 1, 0, 0, 1, 0) ) , .id = "grp")survdiff(formula = Surv(t, m==1) ~ grp, data = data)## Call:## survdiff(formula = Surv(t, m == 1) ~ grp, data = data)## ## N Observed Expected (O-E)^2/E (O-E)^2/V## grp=1 6 4 2.57 0.800 1.62## grp=2 6 3 4.43 0.463 1.62## ## Chisq= 1.6 on 1 degrees of freedom, p= 0.2
From above we can find the Expected column corresponds to the expected value we calculated, and the Observed column represents the observed value, and the (O-E)^2/V column represents the test statistic where the V is the variance of it. And both of them show the same chisq value and p-value.
For now perhaps you have a bette understanding of logrank test like me after going through the whole computation process.
]]>
<p>The logrank test is the most commonly used statistical test in clinical trials to compare the survival distributions in different treatment groups. We usually just use the logrank results to test whether there is a difference between two survival curves. But what does this difference mean?</p>
Quality control of SDTM using the sdtmchecks packagehttp://www.bioinfo-scrounger.com/archives/qc_sdtmchecks/2024-06-19T13:09:02.000Z2024-06-19T13:25:29.419Z如果想对SDTM有个快速全面的check,Pinnacle 21毫无疑问是首选,其能帮助我们对SDTM进行CDISC标准验证,发现data quality issue以及mapping不合理的之处。
若需要一些personalized check过程或者说company-specific check,那么sdtmchecks R包能提供一些非常有用的支持。就像sdtmchecks包介绍中所说的,其并不是想囊括所有SDTM check rules,也不是P21 data validation的复制替代品,其主要是想提供一个一般化且可操作并且有意义的data check。
# Subset to checks that should work OK for most datasetsmetads = sdtmchecksmeta %>% filter(check %in% c("check_ae_aedecod", "check_ae_aetoxgr", "check_ae_dup", "check_cm_cmdecod", "check_cm_missing_month", "check_dm_age_missing", "check_dm_usubjid_dup", "check_dm_armcd" ))myreport <- run_all_checks(metads = metads, verbose = TRUE)
]]>
<p>如果想对SDTM有个快速全面的check,Pinnacle 21毫无疑问是首选,其能帮助我们对SDTM进行CDISC标准验证,发现data quality issue以及mapping不合理的之处。</p>
Estimated LS-means from Multiple Imputation of mice using emmeans packagehttp://www.bioinfo-scrounger.com/archives/mice-lsmean-emmeans/2024-05-27T14:30:41.000Z2024-05-29T07:25:12.347ZContinue with the question in the previous article (Multiple Imputaton - Linear Regression in R), where we just discussed how to compute the pooled coefficients of ANCOVA using mice package but left out the Ls-means and hypothesis test. Luckly I find out that emmeans package have wrapped this process inside so we can use it to obtain the pooled Ls-means estimation and p-value straightforward wihout pool function of mice.
Supposed that we have fitted the ANCOVA for imputed datasets and get the fitted models mods for each imputation here. Then I will use the emmeans::emmeans() function to estimate the ls-means, which is not the indivival estimate for each imputation but rather the pooled one. The pool process remains to use the Rubin's Rules.
If we want to validate the above result using SAS procedure, we must first export the csv from the low_imp_res dataframe that we created in the last article.
write.csv(low_imp_res, file = "./low_imputed.csv", row.names = FALSE, na = "")
If we want to validate the above result using SAS procedure, we must first export the csv from the low_imp_res dataframe that we created in the last article. And than fit the ANCOVA model with proc mixed to obtain the ls-means estimate(lsm and diff) for each imputation. Finally use proc mianalyze to pool the results of all imputation for ls-means (comb_lsm) and difference (comb_diff) within two groups.
proc import datafile="&_projpth.\02_Raw Data\low_imputed.csv" out=low_imp(where=(imp ne 0)) dbms=csv replace; getnames = yes;run;ods output lsmeans=lsm diffs=diff; proc mixed data=low_imp; by imp; class trt / ref=first; model week8=basval trt /ddfm=kr; lsmeans trt / cl pdiff diff;run;proc sort data=lsm; by trt; run;ods output ParameterEstimates=comb_lsm; proc mianalyze data=lsm; by trt; modeleffects estimate; stderr stderr;run;ods output ParameterEstimates=comb_diff; proc mianalyze data=diff; by trt _trt; modeleffects estimate; stderr stderr;run;
The SAS output can be seen below.
sas_mi_pool
We can observe that the estimate and SE from SAS are consistent with R, but there is a significant discrepancy in DF (degress of freedom). In R df is 197 while in SAS it is 3.94E6. That's because there are different methods to calculate the the df, an older one is used in SAS and adjusted version is used in the mice package. That will also lead to different p-values. For more details can be found in Degrees of Freedom and P-values of Rubins-Rules.
Someone may be curious how to calculate the df in mice and SAS. Let’s repeat the process of calculation using the formulas in above link. The specific formulas are not shown here, which is very easy to understand. So I just convert them as the R code.
The older method used in SAS to calculate the df for the t-distribution is defined as (Rubin (1987), Van Buuren (2018)). The specific formulas are not shown here, which is very easy to understand. So I just convert them as the R code. From below, the lambda can be derived from the between (Vb) and total (Vt) missing data variance, and the m represents the number of imputed datasets. The rounded value is 3.94E6 that is equal to the results in SAS.
As the above df is too larger for the pooled result, compared to the dfs in each imputed dataset, which is inappropriate. Barnard and Rubin (1999) adjusted this df by using a new formula (See formula 9.9 in that article.). We should compute the Observed df (df_obs) and then adjusted df (df_adj) where n represents the sample size in the imputed datasets, and k the number of parameters in the fitted model (in my case, there are 3 parameters).
Finally we can look at the df for the pooled estimates from pool_res using pool() function. The df for trt2 term is about equal to our computation, as seen below.
The df calculation for pooling process in emmeans package for mina class has kept the consistency with mice package, using the the Barnard-Rubin adjustment for small samples (Barnard and Rubin, 1999) that mentioned in the pool() documents, see https://github.com/rvlenth/emmeans/issues/494. Thus we can get the same df in either the mice or emmeans packages. All above results have been updated.
]]>
<p>Continue with the question in the previous article (<a href="https://www.bioinfo-scrounger.com/archives/mi_mice/">Multiple Imputaton - Linear Regression in R</a>), where we just discussed how to compute the pooled coefficients of ANCOVA using <code>mice</code> package but left out the Ls-means and hypothesis test. Luckly I find out that <code>emmeans</code> package have wrapped this process inside so we can use it to obtain the pooled Ls-means estimation and p-value straightforward wihout <code>pool</code> function of <code>mice</code>.</p>
Multiple Imputaton - Linear Regression in Rhttp://www.bioinfo-scrounger.com/archives/mi_mice/2024-05-20T13:47:37.000Z2024-05-21T08:01:14.186ZWe have discussed the multiple imputation in non-monotone pattern of missingness in the article of Understanding Multiple Imputation in SAS, and sort out how to implement it in SAS. While here, I would like to learn how to use linear regression in multiple imputation to deal with monotone pattern data in R.
In R, we can use the mice package (Multiple Imputation with Chained Equations) to perform multiple imputation where there is an option for the linear regression method.
The chained equations is a variation of a Gibbs Sampler (an MCMC approach) that iterates between drawing estimates of missing values and estimates of parameters for distribution of the variable (both conditional on the other variables).
And what's the linear regression imputation and what are the advantages and disadvantages of it? Please see the below explaination from this article (Multiple Imputation).
In regression imputation, the existing variables are used to predict, and then the predicted value is substituted as if an actually obtained value. This approach has several advantages because the imputation retains a great deal of data over the listwise or pairwise deletion and avoids significantly altering the standard deviation or the shape of the distribution. However, as in a mean substitution, while a regression imputation substitutes a value predicted from other variables, no novel information is added, while the sample size has been increased and the standard error is reduced.
As shown above, this is a long format data set, including few variables, and the meaning of them is straightforward to understand literally. In order to meet the mice functions, I will first convert it to wide format with separte variables for each time points (week1 - week8).
Then let's have a look at the missing pattern of this example.
low_wide %>% select(basval, week1, week2, week4, week8) %>% mutate(across(1:5, function(x) { if_else(is.na(x), ".", "X") })) %>% group_by_all() %>% count(name = "Freq") ## # A tibble: 4 × 6## # Groups: basval, week1, week2, week4, week8 [4]## basval week1 week2 week4 week8 Freq## <chr> <chr> <chr> <chr> <chr> <int>## 1 X X . . . 4## 2 X X X . . 3## 3 X X X X . 9## 4 X X X X X 184
Besides you can also use the mice::md.pattern(low_wide) function to display the missing patterns that is similar as the above outputs.
As shown above, the 'X' marks indicate that the data point in a certain visit is completed and '.' marks indicate the data is fully missing. So we can suppose that the example data is monotonic type and missingness is MAR for later analysis purposes.
Now we will generate 5 imputed datasets via mice function by setting m=5 and method = 'norm.predict' (called Linear regression through prediction).
If you would like to check if the missingness are all completed, via low_imp$imp. And you can also specify which imputed datasets to use via setting action argument. The action = 0 will return the orginal dataset with missing values, and action = 1 corresponds to the first imputed datasets.
low_imp_1 <- complete(low_imp, action = 1)
And maybe we'd like to have all imputed datasets in a long format that will be easy to handle and analyze at some point.
low_imp_res <- complete(low_imp, action = "long", include = TRUE)
If you already have an imputed dataset with long format from another imputation method, mice::as.mids() would be a helpful function that can convert it to an object with mids class for the further analysis in mice. The mids class should contains the orginal data as well as imputed datasets with .imp and .id columns inside.
mids <- as.mids(low_imp_res)
The second step is to fit the ANCOVA model for week8 time point with treatment (trt) as independent variable, change from baseline in week 8 (week8) as response variable, and baseline (basval) as covariates.
mods <- with( low_imp, lm(week8 ~ basval + trt))
The mods is an object with mira class that contains the call and fitted model object for each one of the imputations.
We can see the pooled estimations like coefficient and standard error in the above output, which is obtained by coef() and vcov() functions from pooled models such as lm here. The estimated treatment coefficient is around -1.73 with a standard error around 0.67.
Actually the coefficient and standard error are not the final results we would like to display in the clinical report. We should get the LS-means estimation for each group, and do contrast between two groups and hypothesis test. That will be discussed in the next topic.
]]>
<p>We have discussed the multiple imputation in non-monotone pattern of missingness in the article of <a href="https://www.bioinfo-scrounger.com/archives/mi_sas/">Understanding Multiple Imputation in SAS</a>, and sort out how to implement it in SAS. While here, I would like to learn how to use linear regression in multiple imputation to deal with monotone pattern data in R.</p>
使用rtables生成Time-To-Event汇总表http://www.bioinfo-scrounger.com/archives/rtables_tte_summary_table/2024-04-06T15:28:01.000Z2024-04-12T08:33:02.487Z在临床试验中,通常使用SAS来完成统计分析和生成图表,但我们不应该只局限于一种编程方法,而且这个所用的编程语言SAS并不是开源的。毫无疑问SAS能完成的事情,R和Python同样能做;但有些R和Python能做的,SAS却很难完成,我想这就是开源和不开源的区别。
]]>
<p>在临床试验中,通常使用SAS来完成统计分析和生成图表,但我们不应该只局限于一种编程方法,而且这个所用的编程语言SAS并不是开源的。毫无疑问SAS能完成的事情,R和Python同样能做;但有些R和Python能做的,SAS却很难完成,我想这就是开源和不开源的区别。</p>
Common Survival analysis of Oncology trials in Rhttp://www.bioinfo-scrounger.com/archives/survival-oncology-r/2024-03-05T14:17:34.000Z2024-04-29T07:11:53.328ZTime-to-event endpoints are widely used in oncology trials, such as OS and PFS. And survival analysis is a common method for estimating time-to-event endpoints. In this blog, I’d like to make a note of how to summarize the essential results for survival analysis in oncology trials in R and also compare them with SAS.
Normally we will show the primary analyses, like below:
Descriptive statistics of the number of events and censors.
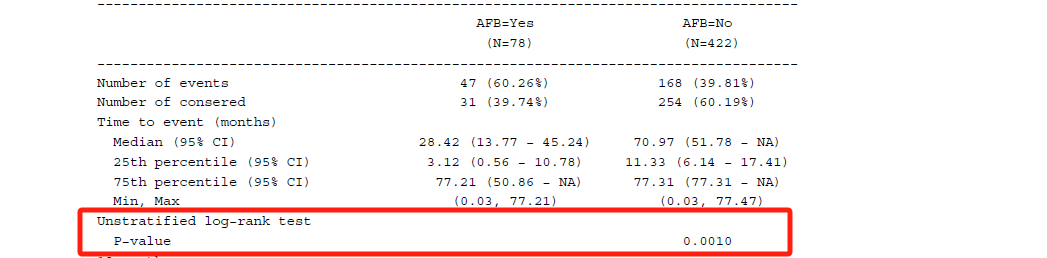
Median (and 25th, 75th percentile) survival time from Kaplan-Meier estimate, along with 95% CI that will be calculated via Brookmeyer and Crowley methodology using log-log transformation.
Survival rate at each time-point of interest from Kaplan-Meier estimate, along with 95% CI that will be calculated via Greenwood formula using log-log transformation.
Hazard Ratio or stratified Hazard Ratio, along with 95% CI from Cox proportional hazards (PH) model, adding Efron approximation for ties handling.
P-value of log-rank test or stratified log-rank test.
Above are the most prevalent survival analysis methods in the Statistical Analysis Plan (SAP) for oncology trials. Let's see how to implement them in R.
In this blog, I will use the example data from the Worcester Heart Attack Study (https://stats.idre.ucla.edu/sas/seminars/sas-survival/) with 500 subjects, which has been wrapped in stabiot R package. And you can find the description of all columns in ?whas500 after installing the package, like devtools::install_github("kaigu1990/stabiot").
library(survival)library(stabiot)data("whas500")
If we want to compare the survival time between the subjects with and without atrial fibrillation, we should first convert the AFB variable to a factor.
In the Surv() function, the event variable takes on the value 1 for events and 0 for censoring, which is in contrast to SAS. And the conf.type = "log-log" tells the function to estimate the CI of median or other percentiles via Brookmeyer and Crowley methodology using log-log transformation because the default argument is conf.type = "log".
Then we can use summary() to see more detail or obtain the median survival time.
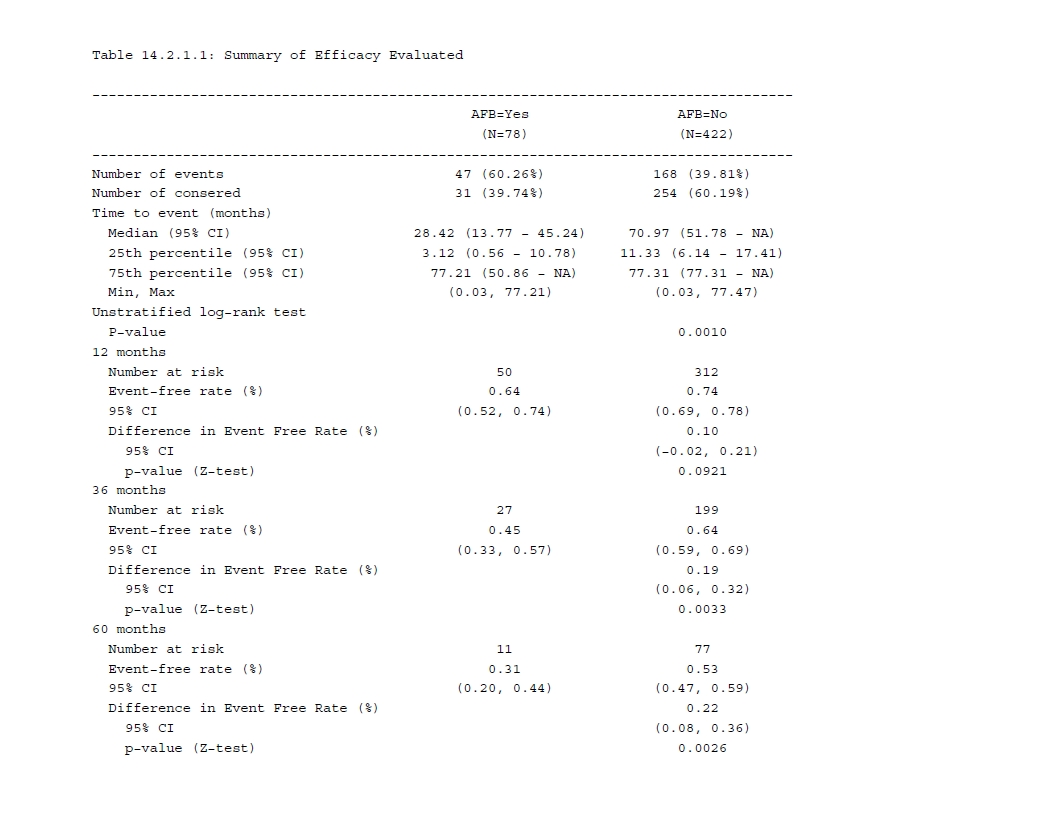
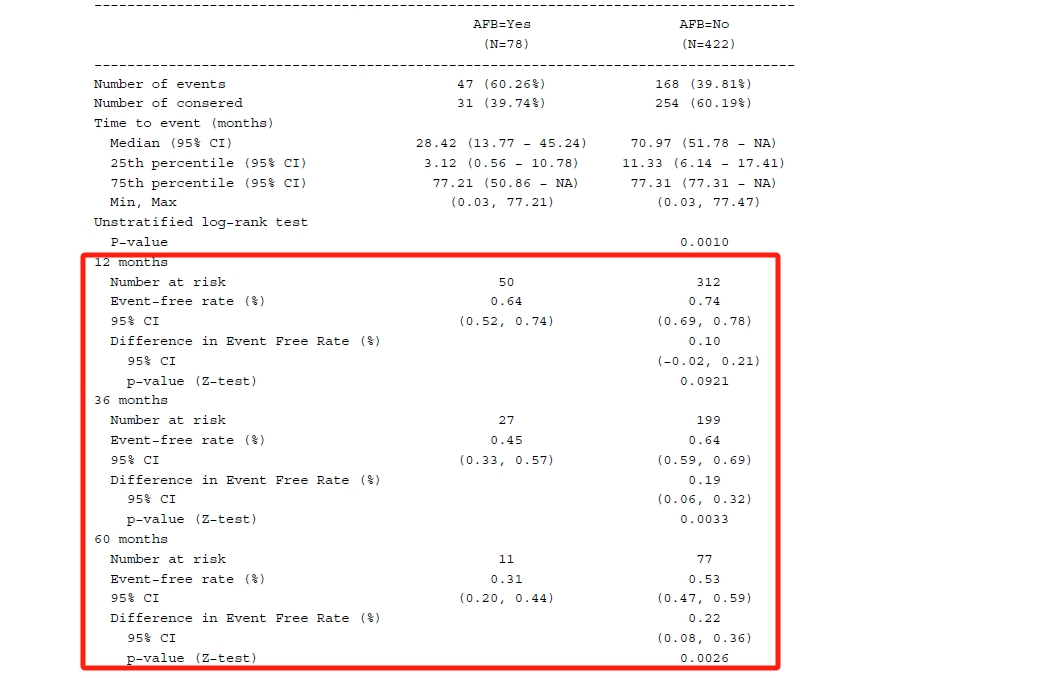
print(summary(fit_km), digits = 4)# median survival time with CIsummary(fit_km)$table## records n.max n.start events rmean se(rmean) median 0.95LCL 0.95UCL## AFB=1 78 78 78 47 35.86989 3.821604 28.41889 13.76591 45.24025## AFB=0 422 422 422 168 48.63073 1.714196 70.96509 51.77823 NA
The n.risk column gives us the number of subjects who are still in the risk condition at specific time points. The n.event column demonstrates the number of events that occurred at the time. And survival column tells us the survival rate from the KM estimate and the last two columns are the corresponding CI.
In addition, you may be interested in the difference rate and corresponding CI between groups with and without AFB. Now that you know the rate and SE for two groups seperately, thus the difference rate and difference SE can be simply calculated. Afterwards for CI calculation, utilize the qnorm() function as follows.
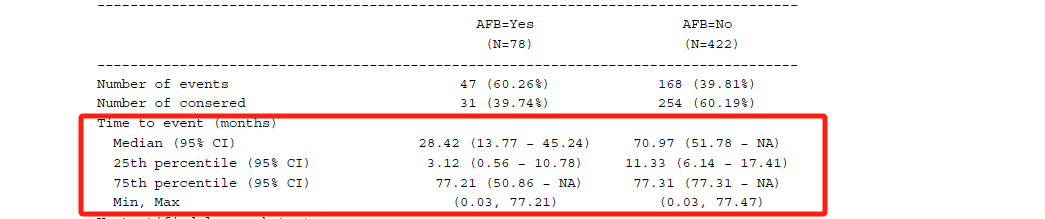
The Log-rank test is a non-parametric test for comparing the survival function across two or more groups where the null hypothesis is that the groups's survival functions are the same. It can be calculated via survminer::surv_pvalue() function with method = "log-rank" for survfit object, or survival::survdiff() function with rho = 0. Both are part of the default set, so you don't need to define them explicitly. Let me show them separately, as shown below.
If you would like to know what is the Log rank test, this article (Log Rank Test) can be for your reference.
Cox PH model
As we will know, the Cox regression model is a semi-parametric model since it makes no assumption about the distribution of the event times that is similar to the KM method of non-parametric, but it relies on a partial likelihood estimation that is partially defined parametrically. Before fitting the Cox model, we should make sure the proportional hazard assumption is met. And more details can be seen at http://www.sthda.com/english/wiki/cox-model-assumptions.
Let's go on the whas500 example data. If you want to estimate the hazard ratio comparing those two groups and also specify the efron approximation for tie handling, the survival::coxph() function can be used for fitting Cox PH models simply.
fit_cox <- coxph(Surv(LENFOL, FSTAT) ~ AFB, data = dat, ties = "efron")## Call:## coxph(formula = Surv(LENFOL, FSTAT) ~ AFB, data = dat, ties = "efron")## ## n= 500, number of events= 215 ## ## coef exp(coef) se(coef) z Pr(>|z|) ## AFB0 -0.5397 0.5829 0.1654 -3.263 0.0011 **## ---## Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1## ## exp(coef) exp(-coef) lower .95 upper .95## AFB0 0.5829 1.716 0.4215 0.8061## ## Concordance= 0.537 (se = 0.014 )## Likelihood ratio test= 9.58 on 1 df, p=0.002## Wald test = 10.64 on 1 df, p=0.001## Score (logrank) test = 10.9 on 1 df, p=0.001
The Cox results can be interpreted as follows: - The coef is the coefficient, and z is the Wald statistic value that corresponds to the rate of coef to its standard error (se(coef)). - Hazard ratio (HR) corresponds to the exp(coef), which is comparing the current level to the reference level. So the HR of 0.58 indicates that the subjects without AFB have 0.58 less hazard or risk compared to those who have AFB. In other words, if the event occurred in 20% of the no AFB group, it would occur in 8.4% (20% - (20% x 0.58)) of the AFB group, which means the no AFB can reduce the hazard of deaths by 0.58. - The HR confidence interval is also provided, with lower 95% bound of 0.4215 and upper 95% bound of 0.8061. - There are three alternative tests for overall significance of the model: likelihood-ratio test, Wald test, and score log-rank statistics. And as we know, log-rank test is a special case of Cox model, which means it equals a univariate Cox regression (only considering treatment). So we can calculate the log-rank p-value from 'survdiff()` as well for the Cox model.
Stratified Log-rank test and Cox PH model
The stratified log-rank test is commonly used for randomized clinical trials when there are baseline factors that may be related to the treatment effect.
The stratified log-rank test is the log-rank test that accounts for the difference in prognostic factors between the two groups. Specifically, we divide the data according to the levels of the significant prognostic factors and form a stratum for each level. At each level, we arrange the survival times in ascending order and calculate the observed number of events, expected number of events, and variance at each survival time as we would in the regular log-rank test. (Chapter 23 - An Introduction to Survival Analysis)
To implement the stratified log-rank test, simply include the strata() within the survival model formula as follows.
Regarding the stratified Cox model, it can be used if there are one or more predictors that don’t satisfy the proportional hazard assumptions. In other words, the proportional hazard is violated.
It can also be performed using coxph along with strata() function, the same as stratified log-rank test.
Above is a summary of common survival analyses in R. In the next step, I would like to wrap these functions into one or two functions and specify the print method so that we can simply use them to compare with SAS.
]]>
<p>Time-to-event endpoints are widely used in oncology trials, such as OS and PFS. And survival analysis is a common method for estimating time-to-event endpoints. In this blog, I’d like to make a note of how to summarize the essential results for survival analysis in oncology trials in R and also compare them with SAS.</p>
Evaluation of Best Overall Response per RECIST in Rhttp://www.bioinfo-scrounger.com/archives/bor_recist/2024-02-28T16:51:20.000Z2024-02-28T16:54:09.678ZThe Best Overall Response (BOR) is a very common evaluation of efficacy in oncology trials. Usually, it is defined as the best response among all time-point responses from the treatment start until the first disease progression, in the order of CR, PR, SD, PD, and NE per RECIST 1.1. For non-randomized trials, BOR is not only the best among all responses but also requires confirmation for CR and PR to ensure the result is not a measurement error. More details can be found in the RECIST 1.1 document, which I will not expand on here.
Although there are lots of blogs on Google that will tell you how to derive BOR in SAS, only a few people will use R to do so. This article is to talk about how to implement BOR with or without confirmation in R.
Best Overall Response without confirmation
Firstly, let's look at the programming logic for BOR without confirmation.
Set to complete response (CR) if one CR exists.
Set to partial response (PR) if one PR exists .
Set to stable disease (SD) if one SD exists, which meets the minimum requirement for SD duration from treatment (or randomization) start to the date of the response.
Set to progressive disease (PD) if one PD exists.
Set to not estimable (NE) if only NE exists or the response cannot meet minimum SD duration criteria.
Afterwards, you can select the best response above for each subject as the BOR.
Actually, we don't consider the scenario where the CR is followed by PR, but we must consider the scenario where subsequent response is not sequential. And we also need to consider how many NEs are acceptable between response and confirmatory response.
Thus the programming logic for confirmed BOR can be summarized as following:
Set to complete response (CR) if there is one confirmatory CR at least a minimum number of days (e.g., 28 days) later, all responses between the two should only be "CR" or "NE", and there are no more than a maximum NE (e.g., one NE) between two responses.
Set to partial response (PR) if there is one confirmatory CR or PR at least a minimum number of days (e.g., 28 days) later, all responses between the two should only be are "CR", "PR" or "NE", and there are no more than a maximum NE (e.g., one NE) between two PR/CR responses.
Set to stable disease (SD) if there is one CR, PR or SD that meets the minimum requirement for the duration from treatment (or randomization) start to the date of that response.
Set to progressive disease (PD) if one PD exists.
Set to not estimable (NE) if there is at least one CR, PR, SD, NE.
And then like the unconfirmed BOR, you can select the best response above for each subject as the confirmed BOR.
How to implement it in R
I have created a function in stabiot R package following the rules we discussed above. For example, let's try it using the derive_bor() function as shown below. More detials can be found in ?derive_bor.
Suppose that we want to calculate the BOR without confirmation and the SD duration is set to 4 weeks, only we simply need to specify ref_start_window = 28.
derive_bor(data = adrs, ref_start_window = 28)## # A tibble: 7 × 8## USUBJID ADTC AVALC ADT TRTSDT PARAMCD PARAM AVAL## <chr> <chr> <chr> <date> <date> <chr> <chr> <dbl>## 1 1 2020-02-01 CR 2020-02-01 2020-01-01 BOR Best Overall Response 1## 2 2 2020-03-13 CR 2020-03-13 2019-12-12 BOR Best Overall Response 1## 3 4 2020-01-01 PR 2020-01-01 2019-12-30 BOR Best Overall Response 2## 4 5 2020-01-01 PR 2020-01-01 2020-01-01 BOR Best Overall Response 2## 5 6 2020-02-16 CR 2020-02-16 2020-02-02 BOR Best Overall Response 1## 6 7 2020-02-16 CR 2020-02-16 2020-02-02 BOR Best Overall Response 1## 7 8 2020-02-16 PD 2020-02-16 2020-02-01 BOR Best Overall Response 4
Suppose that we want to calculate the BOR with confirmation and the SD duration is set to 4 weeks, and the interval of two responses is set to 28 days, we simply need to add ref_interval = 28 and confirm = TRUE
The above all are my summries for BOR calculation. If there is any problem or error, please email me to let me know, or leave your issues in the https://github.com/kaigu1990/stabiot/issues.
At the very least, I'd like to appreciate the admiral R package, I have learned more programming skills for BOR calculation from admiral::derive_extreme_event().
]]>
<p>The Best Overall Response (BOR) is a very common evaluation of efficacy in oncology trials. Usually, it is defined as the best response among all time-point responses from the treatment start until the first disease progression, in the order of CR, PR, SD, PD, and NE per RECIST 1.1. For non-randomized trials, BOR is not only the best among all responses but also requires confirmation for CR and PR to ensure the result is not a measurement error. More details can be found in the RECIST 1.1 document, which I will not expand on here.</p>
Response Rate and Odd Ratio in R and SAShttp://www.bioinfo-scrounger.com/archives/orr_odds_ratio/2024-01-22T14:40:49.000Z2024-04-22T08:33:28.180ZAs we know, the objective response rate (ORR) is used as a key endpoint to demonstrate the efficacy of a treatment in oncology and is also valuable for clinical decision making in phase I-II trials, especially in single-arm trials.
The advantage of the ORR is that it can be assessed earlier than PFS/OS, and in smaller samples. In general, we will assume that the response rate follows the binomial distribution, so naturally we will consider the ORR as a binomial response rate, and the Clopper-Pearson method is frequently used to estimate the two-sided 95% confidence interval (CI). If you would like to control the confounding factors in the stratified study design, the Cochran-Mantel-Haenszel (CMH) test provides a solution to address these needs.
How about the odds ratio (OR)? It is a measure of the association between an exposure and an outcome. So it can be regarded as the odds of the outcome occurring in a particular exposure compared to the odds in the absence of that exposure. Thus, we can use it to assess the ORR between the treatment and control groups in RCT trials in combination with a 95% binomial response rate as presented in reports. More details can be found in Explaining Odds Ratios.
ORR and OR in SAS
Firstly, let's see how to use proc freq in SAS to obtain the ORR rate with Clopper-Pearson (Exact) CI and the odds ratio with and without stratification. Imaging we have an example of data with columns like TRTPN(1/2), ORR(1 for subjects with ORR and 0 without ORR), Strata1(A/B) and the count number.
data dat; input TRTPN ORR Strata1 $ Count @@; datalines;1 1 A 8 1 1 B 121 2 A 17 1 2 B 132 1 A 13 2 1 B 92 2 A 20 2 2 B 8;run;
Then use the tables statement with binomial to compute the CI of ORR. The level="1" binomial option can help you compute the proportion for subjects with events, which means the CI corresponds to the ORR event. And the exact biomial can compute the Clopper-Pearson CI as you need.
Before the stratification analysis, let's see the common odds ratio without any stratified factors. The option chisq requests chi-square tests and measurements, and relrisk displays the odds ratio and relative risk with asymptotic Wald CI by default.
And then let's see how to use CMH as the statistical method in proc freq to obtain the association statistics, p-value of Cochran-Mantel-Haenszel test, adjusted odds ratio by Strata1 variable and corresponding CI.
Now that we have seen the example of the proc freq used to compute the odds ratio with and without stratification, let's have a look at how to use the logistic regression proc logistic to do it.
proc logistic data=dat; weight count; class TRTPN / param=ref ref=last; model ORR(event='1')=TRTPN;run;
And the stratification analysis by logistic as shown below.
proc logistic data=dat; freq count; class TRTPN Strata1 / param=ref ref=last; strata Strata1; model ORR(event='1')=TRTPN;run;
However, we can see there is a little difference between proc freq and the logistic regression method of odds ratio. The same condition occurs in R as well.
ORR and OR in R
Now let's jump into the R section, how can we handle the same analysis in R?
First of all, I want to recommend the tern R package, which focuses on clinical statistical analysis and provides serveral helpful functions. More details can be found in the tern package document.
I create an example data set similar to the one shown above, which includes the same columns but is not the counted table. The columns of strata1 - strata3 represent three stratified factors.
Regarding the unstratification analysis of odds ratio, we can use DescTools::OddsRatio() function, or logistic regression using glm() with logit link. Below is the code to get the odds ratio and corresponding Wald CI using OddsRatio() function.
And the glm() function also can get the same results as shwon below.
fit <- glm(orr ~ trtpn, data = dta, family = binomial(link = "logit"))exp(cbind(Odds_Ratio = coef(fit), confint(fit)))## Odds_Ratio 2.5 % 97.5 %## (Intercept) 0.7857143 0.4450719 1.369724## trtpn1 0.8484848 0.3811997 1.879735
Regarding the unstratification analysis of odds ratio, there are two ways that I have found for computing it. One is Cochran-Mantel-Haenszel chi-squared test using mantelhaen.test() function, and another is conditional logistic regression survival::clogit() function with strata usage for stratification analysis. Let's have a look at the specific steps.
Assuming that we want to consider three stratified factors in our CMH test, we'd better to pre-process data properly before we pass on to mantelhaen.test function. Because this function has certain requirement for the input data format.
# pre-processdf <- dta %>% count(trtpn, orr, strata1, strata2, strata3)tab <- xtabs(n ~ trtpn + orr + strata1 + strata2 + strata3, data = df)tb <- as.table(array(c(tab), dim = c(2, 2, 2 * 2 * 2)))# CMH analysismantelhaen.test(tb, correct = FALSE)## Mantel-Haenszel chi-squared test without continuity correction## data: tb## Mantel-Haenszel X-squared = 0.40574, df = 1, p-value = 0.5241## alternative hypothesis: true common odds ratio is not equal to 1## 95 percent confidence interval:## 0.3376522 1.7320849## sample estimates:## common odds ratio ## 0.7647498
PS. If we only use one stratification like strata1, the same result as SAS proc freq we can get here. Besides you can also use vcdExtra::CMHtest to compute the p-value of CMH, but if you want to obtain the same p-value used in SAS, a modification has to be made to the vcdExtra library. Refer to this github issue: https://github.com/friendly/vcdExtra/issues/3.
And then how to implement it using conditional logistic regression, just add the strata in the formula.
Above all, here is my brief summary for the statisical analysis of ORR and odds ratio in R and SAS. And CMH is also a widely used method to test the association between treatment and binary outcome when you want to consider the stratification factors. Lastly, a question remain unanswered: why do we obtain different results from the logistic regression compared to the CMH test when we apply them to compute the the stratified odds ratio. I'm looking for how to respond to it.
]]>
<p>As we know, the objective response rate (ORR) is used as a key endpoint to demonstrate the efficacy of a treatment in oncology and is also valuable for clinical decision making in phase I-II trials, especially in single-arm trials.</p>
Hypothesis testing of MMRMhttp://www.bioinfo-scrounger.com/archives/mmrm_hypothesis/2023-12-21T13:35:37.000Z2023-12-21T13:37:11.000ZOriginally, I created an issue in CAMIS github asking how to do the hypothesis testing of MMRM in R, especially in non-inferiority or superiority trials. And then I received a reminder that I can get the manual from mmrm package document.
The mmrm package has provided the df_1d() function to do the one-dimensional contrast. So let's start by fitting a mmrm model first with us (unstructured) covariance structure and Kenward-Roger adjustment methods. I also include a linear Kenward-Roger approximation for coefficient covariance matrix adjustment so that R results can be compared with SAS when the unstructured covariance model is selected.
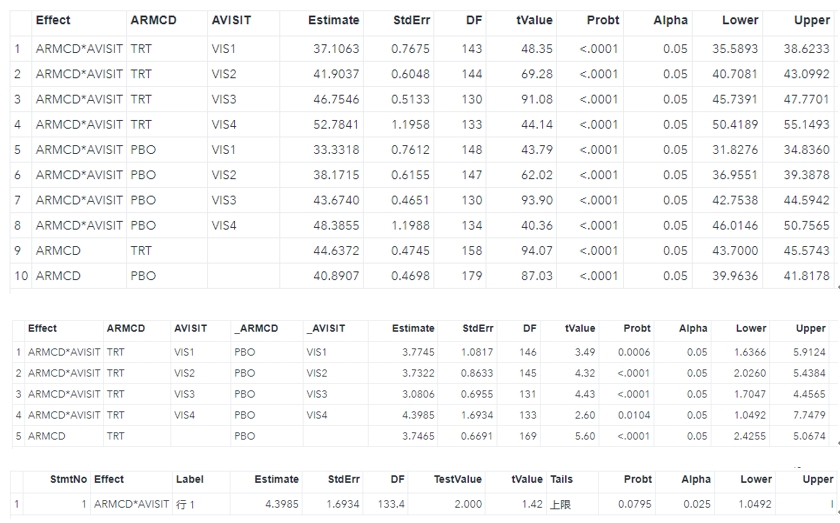
library(mmrm)fit <- mmrm( formula = FEV1 ~ RACE + SEX + ARMCD * AVISIT + us(AVISIT | USUBJID), reml = TRUE, method = "Kenward-Roger", vcov = "Kenward-Roger-Linear", data = fev_data)summary(fit)
Assuming that we aim to compare Race white with Race Asian, the results are as follows.
contrast <- numeric(length(component(fit, "beta_est")))contrast[3] <- 1df_1d(fit, contrast)# same as # emmeans(fit, ~ RACE) %>% contrast() %>% test()
Honestly, I prefer to use the emmeans package to compute estimated marginal means (least-square means), especially when you also want to compute it by visit and by treatment. Because mmrm package sets an object interface so that it can be used for the emmeans package. And emmeans has also built a set of useful functions to deal with common questions. So it’s a good solution to fit the MMRM model by mmrm and do hypothesis testing by emmeans.
A general assumption is that we would like to compute the least-square means first for the coefficients of the MMRM by visit and by treatment. This can be done through emmeans() and confint() functions.
library(emmeans)ems <- emmeans(fit, ~ ARMCD | AVISIT)confint(ems)## AVISIT = VIS1:## ARMCD emmean SE df lower.CL upper.CL## PBO 33.3 0.761 148 31.8 34.8## TRT 37.1 0.767 143 35.6 38.6## ## AVISIT = VIS2:## ARMCD emmean SE df lower.CL upper.CL## PBO 38.2 0.616 147 37.0 39.4## TRT 41.9 0.605 143 40.7 43.1## ## AVISIT = VIS3:## ARMCD emmean SE df lower.CL upper.CL## PBO 43.7 0.465 130 42.8 44.6## TRT 46.8 0.513 130 45.7 47.8## ## AVISIT = VIS4:## ARMCD emmean SE df lower.CL upper.CL## PBO 48.4 1.199 134 46.0 50.8## TRT 52.8 1.196 133 50.4 55.1## ## Results are averaged over the levels of: RACE, SEX ## Confidence level used: 0.95
Naturally we will also want to consider the contrast to see what is the difference between treatment and placebo where the null hypothesis is that treatment minus placebo equals zero. Here the contrast() function will be run. If you want to see the confidence interval of difference, just use confint(contr) that will be fine. PS. You can relevel the order of ARMCD factor in advance, in that case the method=pairwise can reach the same results as well.
contr <- contrast(ems, adjust = "none", method = "revpairwise")contr## AVISIT = VIS1:## contrast estimate SE df t.ratio p.value## TRT - PBO 3.77 1.082 146 3.489 0.0006## ## AVISIT = VIS2:## contrast estimate SE df t.ratio p.value## TRT - PBO 3.73 0.863 145 4.323 <.0001## ## AVISIT = VIS3:## contrast estimate SE df t.ratio p.value## TRT - PBO 3.08 0.696 131 4.429 <.0001## ## AVISIT = VIS4:## contrast estimate SE df t.ratio p.value## TRT - PBO 4.40 1.693 133 2.597 0.0104## ## Results are averaged over the levels of: RACE, SEX
Besides maybe we would like to further assess whether treatment is superior to placebo with a margin of 2. You can utilize the test() function with the null = 2 argument.
test(contr, null = 2, side = ">")## AVISIT = VIS1:## contrast estimate SE df null t.ratio p.value## TRT - PBO 3.77 1.082 146 2 1.640 0.0516## ## AVISIT = VIS2:## contrast estimate SE df null t.ratio p.value## TRT - PBO 3.73 0.863 145 2 2.007 0.0233## ## AVISIT = VIS3:## contrast estimate SE df null t.ratio p.value## TRT - PBO 3.08 0.696 131 2 1.554 0.0613## ## AVISIT = VIS4:## contrast estimate SE df null t.ratio p.value## TRT - PBO 4.40 1.693 133 2 1.416 0.0795## ## Results are averaged over the levels of: RACE, SEX ## P values are right-tailed
In general the common estimations and hypothesis testing of MMRM are all here, which at least I have encountered. In the next step, I want to compare the above results with SAS to see if it can be regarded as additional QC validation. We use the lsmeans statement to estimate least-square means and do superiority testing at visit 4 through lsmestimate statement.
]]>
<p>Originally, I created an <a href="https://github.com/PSIAIMS/CAMIS/issues/41" target="_blank" rel="noopener">issue</a> in <code>CAMIS</code> github asking how to do the hypothesis testing of MMRM in R, especially in non-inferiority or superiority trials. And then I received a reminder that I can get the manual from <code>mmrm</code> package <a href="https://openpharma.github.io/mmrm/main/articles/introduction.html?q=lsmeans#hypothesis-testing" target="_blank" rel="noopener">document</a>.</p>
Contrasts and Hypothesis Tests of emmeanshttp://www.bioinfo-scrounger.com/archives/contrasts_emmeans/2023-12-18T14:09:02.000Z2023-12-18T14:24:21.313ZIn the article Definition of least-squares means (LS means), we have known how to compute the LS mean step by step and how to implement it in the emmeans package that will calculate the estimated mean value for different factor variables and assume the mean value for continuous variables.
In addition, emmeans also contains a set of functions not limited for contrasts and hypothesis testing that are commonly used in clinical trial statistical analysis, such as ANCOVA and MMRM. So the goal of this article is not only to know how to use the emmeans package to answer these questions but also to learn several separate steps for each of them.
Let's start by fitting a model first. Given that I have an example of data fev_data from mmrm package, and then fit a simple ANCOVA model by lm function.
Next, we will create a contrast that is a linear combination of the means. In the following example, the contrast may answer the question of weather the treatment (TRT) produces a significant effect than placebo, like contrast=TRT-PRB.
# TRT vs. PBO: TRT - PBOk <- c(-1, 1)
And then compute the estimation and standard error for the contrast. The basic principle of SE is that the variance of a linear combination of independent estimates is equal to the linear combination of their variances.
est <- tidy(ems) %>% pull(estimate)se <- tidy(ems) %>% pull(std.error)con_est <- con %*% estcon_se <- sqrt(con^2 %*% se^2)
Since we have got the estimation(con_est) and standard error(con_se) above, naturally we can use them to compute the confidence interval and p value following the t distribution with assuming the null hypothesis is TRT - PBO = 0
The same process can be implemented by the emmeans::test() function.
test(contr, null = 2, side = ">", adjust = "none")## contrast estimate SE df null t.ratio p.value## c(-1, 1) 4.83 1.74 132 2 1.625 0.0532## P values are right-tailed
The above is only a simple example of a 1-way ANOVA, so that I can learn and understand it clearly. Actually, other complicated models and contrasts can be processed as well. Through these step by step computations, we can gain deeper thoughts of why we chose the functions, and what's the nature of our computation.
]]>
<p>In the article <a href="https://www.bioinfo-scrounger.com/archives/definition-lsmeans/">Definition of least-squares means (LS means)</a>, we have known how to compute the LS mean step by step and how to implement it in the <code>emmeans</code> package that will calculate the estimated mean value for different factor variables and assume the mean value for continuous variables.</p>
Hexo迁移 - 更换ECS服务器http://www.bioinfo-scrounger.com/archives/hexo_migration/2023-11-26T13:04:05.000Z2023-11-26T13:08:04.179Z趁着最近阿里云双11的优惠活动,我计划更换下博客所在的ECS服务器(其实为了响应消费降级~),咨询了下售前和售后,最终顺利完成迁移,记录一下迁移过程以备后续所需。
]]>
<p>趁着最近阿里云双11的优惠活动,我计划更换下博客所在的ECS服务器(其实为了响应消费降级~),咨询了下售前和售后,最终顺利完成迁移,记录一下迁移过程以备后续所需。</p>
Understanding Mixed Model Repeated Measures (MMRM) in SAS and Rhttp://www.bioinfo-scrounger.com/archives/mmrm_sas_r/2023-10-31T13:08:36.000Z2023-11-01T03:31:15.118ZMixed models for repeated measures (MMRM) is widely used for analyzing longitdinal continuous outcomes in randomized clinical trials. Repeated measures refer to multiple measures taken from the same experimental unit, such as a couple of tests over time on the same subject. And the advantage of this model is that it can avoid model misspcification and provide unbiased estimation for data that is missing completely at random (MCAR) or missing at random (MAR).
As we know, the common primary outcome in randomized trials is often the difference in average (LS mean) at a given timepoint (visit). One way to analyze these data is to ignore the measurements at intermediate timepoints and focus on estimating the outcome at the specific timepoint by ANCOVA, but the data should be complete. If not, sometimes the multiple imputation method is suggested. However in the MMRM model, it's generally thought that utilizing the information from all timepoints implicitly handles missing data. In SAS, it's more efficient to use proc mixed than proc glm to handle missing values, which allows the inclusion of subjects with missing data. And in R, I feel like the 'mmrm' package is more powerful and runs more smoothly than others.
Example data
Here, I take the example data from mmrm package and implement the MMRM using SAS and R, respectively. In this randomized trial, subjects are treated with a treatment drug or placebo, and the FEV1 (forced expired volume in one second) is a measure of how quickly the lungs can be emptied. This measure is repeated from Visit 1 to Visit 4. Low levels of FEV1 may indicate chronic obstructive pulmonary disease (COPD). To evaluate the effect of treatment on FEV1, the MMRM will be used to analyze the outcome with an unstructured covariance matrix reflecting the correlation between visits within the subjects, treatment (treatment drug or placebo), visit and treatment-by-visit as the fixed effects, subject as a randon effect, visit as a repeated measure, and baseline as the covariates.
Implementation
Here, I take the example data from mmrm package and implement the MMRM using SAS and R, respectively. In this randomized trial, subjects are treated with a treatment drug or placebo, and the FEV1 (forced expired volume in one second) is a measure of how quickly the lungs can be emptied. This measure is repeated from Visit 1 to Visit 4. Low levels of FEV1 may indicate chronic obstructive pulmonary disease (COPD).
library(mmrm)data("fev_data")write.csv(fev_data, file = "./fev_data.csv", na = "", row.names = F)
To evaluate the effect of treatment on FEV1, this endpoint measurements can be analyzed using MMRM with an unstructured covariance matrix reflecting the correlation between visits within the subjects, treatment (treatment drug or placebo), visit and treatment-by-visit as the fixed effects, subject as a randon effect, visit as a repeated measure, and race as the covariate.
From above SAS code, we can see that the method option specifies the estimation method as REML. The repeated statement is used to specify the repeated measures factor and control the covariance structure. In the repeated measures models, the subject optional is used to define which observations belong to the same subject, and which belong to the different subjects who are assumed to be independent. The type optional statement specifies the model for the covariance structure of the error within subjects. We also add ddfm=KR in model statement to specify a method for the denominator degrees of freedom (such as Kenward-Rogers here). At least, the LS mean calculated from the lsmeans statement with ci and diff options is also very commonly used. These two options can help us obtain the confidence interval and difference of the LS mean, and the p value if the hypothesis margin is 0.
As for ARMCD*AVISIT in the lsmeans statement that means you would like to get the test of LS means in all combinations of visits. If you try the lsmeans ARMCD, which is identical to the mean of pair-wise visits from the LS means of lsmeans ARMCD*AVISIT.
If you would like to obtain the LS mean of each visit for each group, like the lsm dataset in SAS, you can use the emmeans function from the emmeans package as the mmrm object can be analyzed by the external package.
# emmeans(fit, "ARMCD", by = "AVISIT")emmeans(fit, ~ ARMCD | AVISIT)## AVISIT = VIS1:## ARMCD emmean SE df lower.CL upper.CL## PBO 33.3 0.757 149 31.8 34.8## TRT 37.1 0.764 144 35.6 38.6## ## AVISIT = VIS2:## ARMCD emmean SE df lower.CL upper.CL## PBO 38.2 0.608 150 37.0 39.4## TRT 41.9 0.598 146 40.7 43.1## ## AVISIT = VIS3:## ARMCD emmean SE df lower.CL upper.CL## PBO 43.7 0.462 131 42.8 44.6## TRT 46.8 0.507 130 45.8 47.8## ## AVISIT = VIS4:## ARMCD emmean SE df lower.CL upper.CL## PBO 48.4 1.189 134 46.0 50.7## TRT 52.8 1.188 133 50.4 55.1## ## Results are averaged over the levels of: RACE ## Confidence level used: 0.95
As for the diff dataset from SAS, you can use the pairs function to get identical outputs.
pairs(emmeans(fit, ~ ARMCD | AVISIT), reverse = TRUE, adjust="tukey")## AVISIT = VIS1:## contrast estimate SE df t.ratio p.value## TRT - PBO 3.78 1.076 146 3.508 0.0006## ## AVISIT = VIS2:## contrast estimate SE df t.ratio p.value## TRT - PBO 3.76 0.853 148 4.405 <.0001## ## AVISIT = VIS3:## contrast estimate SE df t.ratio p.value## TRT - PBO 3.11 0.689 132 4.509 <.0001## ## AVISIT = VIS4:## contrast estimate SE df t.ratio p.value## TRT - PBO 4.41 1.681 133 2.622 0.0098## ## Results are averaged over the levels of: RACE
Questions
Why we must include the interaction effect in the model?
I feel like if we use the ANCOVA model and focus on the specific timepoint before the end of the trial, in that case, we can say the treatment effect is the main difference between the treatment and control groups. But in MMRM, we include all timepoints's information. Despite the collection of these intermediate outcomes, the primary outcome is often still the difference at that specific or final timepoint. Thus, it will have a couple of advantages, like improving the power and avoiding the bias of dropout because although the subjects withdraw from the study before the final timepoint, they may still contribute information in the interim. Once all the timepoints are included, the treatment-by-visit also should be added to the model as a consideration when the effect is different in the slopes of outcomes over time.
How to select the covariance structure?
Initially, the unstructured (type=UN) covariance structure allows SAS to estimate the covariance matrix, as the unstructured approach makes no assumption at all about the relationship in the correlations among study visits. As for how to select an appropriate covariance structure, it depends on your understanding of the study and the data you have. Here are also a couple of documents for your reference if you would like to know which structure can be used and how to try and select a more suitable structure. For instance, the lower AIC values suggest a better fit.
]]>
<p>Mixed models for repeated measures (MMRM) is widely used for analyzing longitdinal continuous outcomes in randomized clinical trials. Repeated measures refer to multiple measures taken from the same experimental unit, such as a couple of tests over time on the same subject. And the advantage of this model is that it can avoid model misspcification and provide unbiased estimation for data that is missing completely at random (MCAR) or missing at random (MAR).</p>
mcradds R Packagehttp://www.bioinfo-scrounger.com/archives/mcradds/2023-10-13T02:50:20.000Z2023-10-13T02:58:24.948ZI'm tickled pink to announce the release of mcradds (version 1.0.1) helps with designing, analyzing and visualization in In Vitro Diagnostic trials.
You can install it from CRAN with:
install.packages("mcradds")
or you can install the development version directly from GitHub with:
if (!require("devtools")) { install.packages("devtools")}devtools::install_github("kaigu1990/mcradds")
This blog post will introduce you to package and desirability functions. Let's start loading this package.
library(mcradds)
The mcradds R package is a complement to mcr package and it offers common and solid functions for designing, analyzing, and visualizing in In Vitro Diagnostic (IVD) trials. In my work experience as a statistician for diagnostic trials at Roche Diagnostic, mcr package is an internally built tool for analyzing regression and other relevant methodologies that are also widely used in the IVD industry community.
However, the mcr package focuses on method comparison trials and does not include additional common diagnostic methods but that have been provided in the mcradds. It is intuitive and easy to use. So you can perform statistical analysis and graphics in different IVD trials utilizing the analytical functions.
Estimate the sample size for trials, following NMPA guidelines.
Evaluate diagnostic accuracy with/without reference, following CLSI EP12-A2.
Perform regression method analysis and plots, following CLSI EP09-A3.
Perform bland-Altman analysis and plots, following CLSI EP09-A3.
Detect outliers with 4E method from CLSI EP09-A2 and ESD from CLSI EP09-A3.
Estimate bias in medical decision level, following CLSI EP09-A3.
Perform Pearson and Spearman correlation analysis, adding hypothesis test and confidence interval.
Evaluate Reference Range/Interval, following CLSI EP28-A3 and NMPA guidelines.
Add paired ROC/AUC test for superiority and non-inferiority trials, following CLSI EP05-A3/EP15-A3.
Perform reproducibility analysis (reader precision) for immunohistochemical assays, following CLSI I/LA28-A2 and NMPA guidelines.
Evaluate precision of quantitative measurements, following CLSI EP05-A3.
Please be noted that these functions and methods have not been validated and QC'ed, so I cannot guarantee that all of them are entirely proper and error-free. But I always strive to compare the results to those of other resources in order to obtain a consistent result for them. And because some of them were utilized in my past usual work process, I believe the quality of this package is temporarily sufficient to use.
Let's demonstrate that by looking at a few of examples. More detailed usages can be found in Get started page
Suppose that we have a new diagnostic assay with the expected sensitivity criteria of 0.9, and the clinical acceptable criteria is 0.85. If we conduct a two-sided normal Z-test at a significance level of α = 0.05 and achieve a power of 80%, what should the total sample size be?
The result from sample size function is:
size_one_prop(p1 = 0.9, p0 = 0.85, alpha = 0.05, power = 0.8)#> #> Sample size determination for one Proportion #> #> Call: size_one_prop(p1 = 0.9, p0 = 0.85, alpha = 0.05, power = 0.8)#> #> optimal sample size: n = 363 #> #> p1:0.9 p0:0.85 alpha:0.05 power:0.8 alternative:two.sided
Suppose that you have a wide structure of data like qualData that contains the qualitative measurements of the candidate (your own product) and comparative (reference product) assays. In this scenario, if you’re interested in how to create a 2x2 contingency table, the diagTab() function is a good solution.
And then you can use the getAccuracy() method to compute the diagnostic performance based on the table above.
# Default method is Wilson score, and digit is 4.tb %>% getAccuracy(ref = "r")#> EST LowerCI UpperCI#> sens 0.8841 0.8200 0.9274#> spec 0.8710 0.7655 0.9331#> ppv 0.9385 0.8833 0.9685#> npv 0.7714 0.6605 0.8541#> plr 6.8514 3.5785 13.1181#> nlr 0.1331 0.0832 0.2131
If you want to estimate the reader precision between different readers, reads, or sites, use the APA, ANA and OPA as the primary endpoint in the PDL1 assay trials. Let’s see an example of precision between readers.
data("PDL1RP")reader <- PDL1RP$btw_readertb1 <- reader %>% diagTab( formula = Reader ~ Value, bysort = "Sample", levels = c("Positive", "Negative"), rep = TRUE, across = "Site" )getAccuracy(tb1, ref = "bnr", rng.seed = 12306)#> EST LowerCI UpperCI#> apa 0.9479 0.9260 0.9686#> ana 0.9540 0.9342 0.9730#> opa 0.9511 0.9311 0.9711
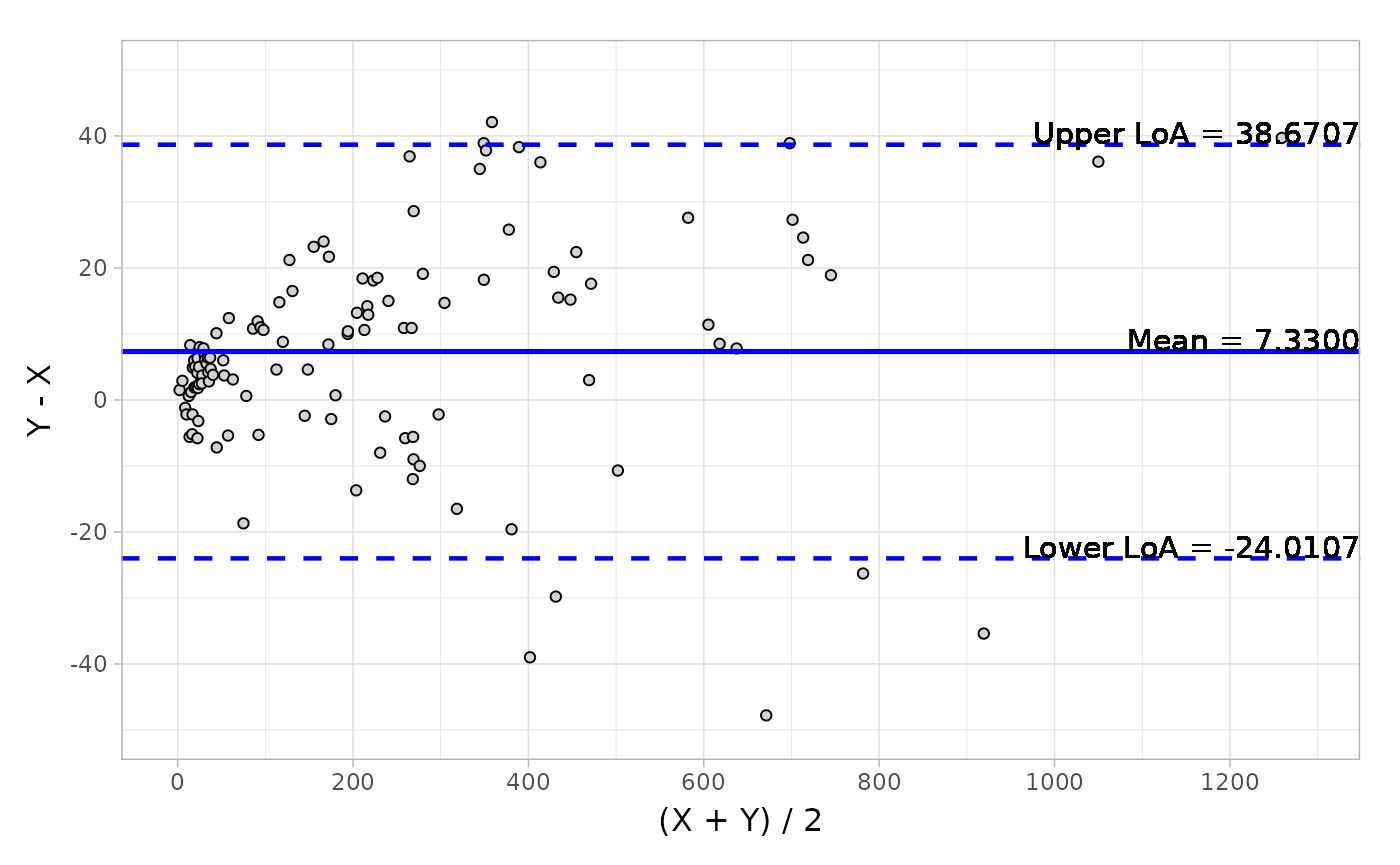
Suppose that in another scenario, you have a wide structure of quantitative data like platelet and would like to do the Bland-Altman analysis to obtain a series of descriptive statistics including, mean, median, Q1, Q3, min, max and other estimations like CI (confidence interval of mean) and LoA (Limit of Agreement).
data("platelet")# Default difference typeblandAltman( x = platelet$Comparative, y = platelet$Candidate, type1 = 3, type2 = 5)#> Call: blandAltman(x = platelet$Comparative, y = platelet$Candidate, #> type1 = 3, type2 = 5)#> #> Absolute difference type: Y-X#> Relative difference type: (Y-X)/(0.5*(X+Y))#> #> Absolute.difference Relative.difference#> N 120 120#> Mean (SD) 7.330 (15.990) 0.064 ( 0.145)#> Median 6.350 0.055#> Q1, Q3 ( 0.150, 15.750) ( 0.001, 0.118)#> Min, Max (-47.800, 42.100) (-0.412, 0.667)#> Limit of Agreement (-24.011, 38.671) (-0.220, 0.347)#> Confidence Interval of Mean ( 4.469, 10.191) ( 0.038, 0.089)
And the visualization of Bland-Altman can be easily conducted by the autoplot method.
object <- blandAltman(x = platelet$Comparative, y = platelet$Candidate)# Absolute difference plotautoplot(object, type = "absolute")
Here is a plot of the data.
Bland-Altman_plot
Based on the output from Bland-Altman, you can also detect the potential outliers using the getOutlier() method.
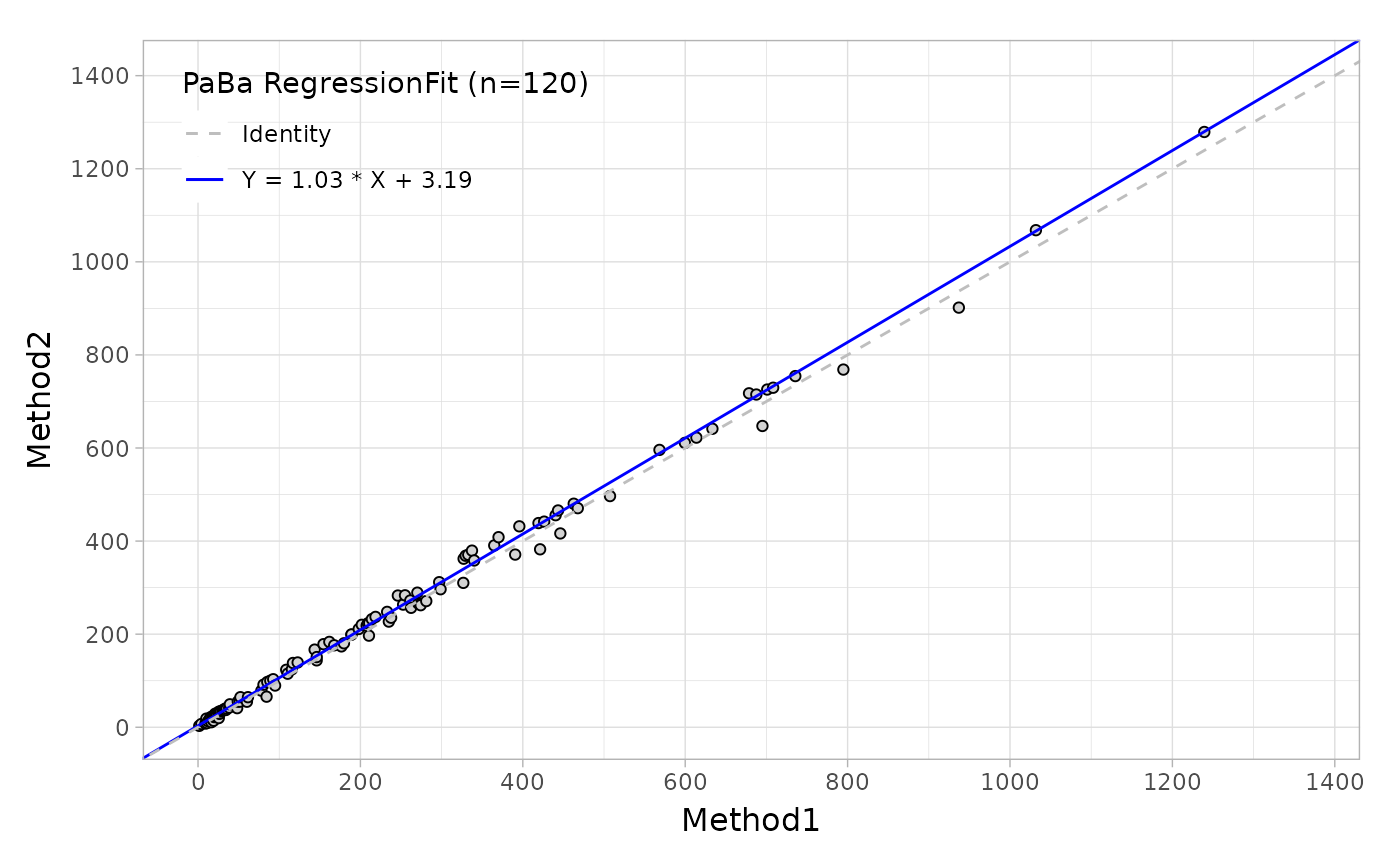
Suppose that you would like to evaluate the regression agreement between two assays with 'Deming' method, you can use the mcreg, this main function is wrapped from mcr package.
# Deming regressionfit <- mcreg( x = platelet$Comparative, y = platelet$Candidate, error.ratio = 1, method.reg = "Deming", method.ci = "jackknife")
Like the Bland-Altman plot, as well as in regression plot, the autoplot function can provide the scatter plot with a fitted line as shown below.
Regression_plot
Based on this regression analysis, you can also estimate the bias at one or more medical decision levels.
Suppose that you want to see if the OxLDL assay is superior to the LDL assay through comparing two AUC of paired two-sample diagnostic assays using the standardized difference method when the margin is equal to 0.1. In this case, the null hypothesis is that the difference is less than 0.1.
data("ldlroc")# H0 : Superiority margin <= 0.1:aucTest( x = ldlroc$LDL, y = ldlroc$OxLDL, response = ldlroc$Diagnosis, method = "superiority", h0 = 0.1)#> Setting levels: control = 0, case = 1#> Setting direction: controls < cases#> #> The hypothesis for testing superiority based on Paired ROC curve#> #> Test assay:#> Area under the curve: 0.7995#> Standard Error(SE): 0.0620#> 95% Confidence Interval(CI): 0.6781-0.9210 (DeLong)#> #> Reference/standard assay:#> Area under the curve: 0.5617#> Standard Error(SE): 0.0836#> 95% Confidence Interval(CI): 0.3979-0.7255 (DeLong)#> #> Comparison of Paired AUC:#> Alternative hypothesis: the difference in AUC is superiority to 0.1#> Difference of AUC: 0.2378#> Standard Error(SE): 0.0790#> 95% Confidence Interval(CI): 0.0829-0.3927 (standardized differenec method)#> Z: 1.7436#> Pvalue: 0.04061
Suppose that you feel like to do the hypothesis test of H0=0.7 not H0=0 with pearson and spearman correlation analysis, the pearsonTest() and spearmanTest() would be helpful.
That's it! That's the mcradds package. More details can be found in the Introduction to mcradds vignette.
]]>
<p>I'm tickled pink to announce the release of <code>mcradds</code> (version 1.0.1) helps with designing, analyzing and visualization in In Vitro Diagnostic trials.</p>
Releasing R Package to CRANhttp://www.bioinfo-scrounger.com/archives/release_cran/2023-10-12T12:30:28.000Z2024-08-27T13:27:53.220ZRecently, I've been developing my R package - mcradds, which will be my first package released to CRAN. To be honest, finishing coding is just the first step for R package development, whereas I feel like the submission to CRAN is the most challenging for me. This blog is to keep track of something I came across during the submission process to help giving me a reminder when I would develop other packages in next steps. If you are a beginner like me, this blog will be beneficial to you as well.
Use usethis::use_release_issue() to generate a listing on the github issue page to advise on a series of recommendations you should finish.
If you don't have a README document already, you should create and render devtools::build_readme() it before releasing. Don't forget to add the install instructions in the README. Keep updating the NEW document as well.
A vignette is necessary that is a long-term guide to your package. Use usethis::use_vignette("my-vignette") to create a default template first, and then you can just follow other mature packages's vignettes, through following the similar structure from them is okay (that's what I'm doing).
In addition, a website like pkgdown is also help for users to know more about your package. These functions from usethis package can help your build it. The usethis::use_pkgdown() function to initial setup, and pkgdown::build_site() to render your site, then usethis::use_pkgdown_github_pages() to deployment your site to github and githun action.
Check the DESCRIPTION clearly
Proofread the title, follow the naming rule, like it should be plain text (no markup), capitalized like a title, and NOT end in a period.
Provide a good description, which is very important.
Check version number, updating manually or using usethis::use_version().
Don't forget to add (copyright holder) role to Authors@R. If you are the only developer, you should add three roles and put "aut", "cre" and "cph" together.
Make sure the license is reasonable and correct.
Add the correct urls following to the CRAN's URL checks, and check with urlchecker::url_check().
Check and list all spell words in inst/WORDLIST automatically with usethis::use_spell_check(). That's a fantastic way—just a one-line command.
At last, run devtools::check() once again to ensure everything is ready.
Releasing Preparation
As usual, I use devtools::check() to double-check all is still well before I want to merge or commit update. But before releasing, you'd better add remote = TRUE and manual = TRUE to run the R CMD check again, like devtools::check(remote = TRUE, manual = TRUE), which will build and check the manual, and perform a number of CRAN incoming checks.
Maybe you will encounter the same problem I had, like a confused warning pdflatex not found! Not building PDF manual. I didn't understand the meaning of this warning at first. I checked all options in R and Rstudio, but that didn't work. Finally, I found that it occurred because I didn't have the pdflatex executive program on this computer!
It's easy to solve the problem if you find it. I chose to install the pdflatex using the solution provided by Yihui Xie, referring to the article https://yihui.org/tinytex/.
Another option is to add some more packages for building PDF vignettes of many CRAN packages.
tinytex:::install_yihui_pkgs()
At last, if it still doesn't work, ensure the path of pdflatex has been added to your PATH environment on the computer.
After R CMD check, you'd better use devtools::check_win_devel() as this checking with r-devel is required by CRAN policy. And make sure your package can be passed through CRAN's win-builder service, which is only for Windows. Another good option is to use rhub::check_for_cran() that is also a service supported by the R Consortium, to check your package.
If this package is the new submission to CRAN, there are currently no downstream dependencies for it. If not, you should do the reverse dependency checks.
After all the above, record comments about the submission to cran-comments.md, and that will be created by the usethis::use_cran_comments() you use at first. There is no need to manually add it.
Submit to CRAN
Once you're satisfied that all issues have been addressed and it's time to submit your package to CRAN, run usethis::use_version() to reach the final version you would like for the first release to CRAN, and then submit using devtools::submit_cran() without any hesitation.
Afterwards, you will receive an email telling you that the package is pending a manual inspection of this new CRAN submission. You will get a response within the next 10 working days, but sometime the feedback is very fast.
If there are some comments from CRAN, respond to any CRAN remarks and double-check everything. Fix what needs to be fixed. If not, write and provide a good reason as you can. Don't forget to add a "Resubmission" section at the top of cran-comments.md to clearly identify that the package is a resubmission, and list the changes that you have made. If you want to explain or clarify something, also can be added inside.
At last, if you receive an email telling your package will be published within 24 hours in the correponding CRAN directory, that means your package have been accepted and released on CRAN. And then you should push it to Github with the new version number. And next, use usethis::use_github_release() to create a new release with tag version on your github, and then update the NEW document as well to illustrate that this is the CRAN release.
Now you can continue increasing the version number to the development version using usethis::use_dev_version(). It makes sense to immediately push to GitHub so that any update will be based on the development version.
Other checking lists for CRAN are also available for reference.
]]>
<p>Recently, I've been developing my R package - mcradds, which will be my first package released to CRAN. To be honest, finishing coding is just the first step for R package development, whereas I feel like the submission to CRAN is the most challenging for me. This blog is to keep track of something I came across during the submission process to help giving me a reminder when I would develop other packages in next steps. If you are a beginner like me, this blog will be beneficial to you as well.</p>
Convert Plots to Editable Format in Rhttp://www.bioinfo-scrounger.com/archives/editable_plots/2023-08-27T12:48:54.000Z2023-08-27T12:52:01.317Z推荐一个R包(officer)可以用于生成editable图片在PPT中。这里的editable是指图片中每个元素包括散点、X/Y轴、标签都能修改,常用于图片的再修饰
]]>
<p>推荐一个R包(<code>officer</code>)可以用于生成editable图片在PPT中。这里的editable是指图片中每个元素包括散点、X/Y轴、标签都能修改,常用于图片的再修饰</p>
<p>参考于:<a href="https://ardata-fr.github.io/officeverse/officer-for-powerpoint.html" target="_blank" rel="noopener">Chapter 5 officer for PowerPoint</a></p>
Multiple Imputation in Non-inferiority and Superiority Trialshttp://www.bioinfo-scrounger.com/archives/mi_ni_sp_trials/2023-08-20T13:06:58.000Z2023-08-20T13:10:29.580ZIn the previous article (Understanding Multiple Imputation in SAS), we talked about how to implement multiple imputation in the SAS procedure to compare the difference between the treatment and placebo groups. Let's look at how to do it in non-inferiority and superiority trials, which differ from common use.
In terms of the ANCOVA model, if you would like to add the margin of non-inferiority and superiority, you can just use the lsmestimate statement with testvalue=2 when the margin is 2. Whereas for multiple imputation you can't just add this statement in the analysis step, you should define this margin in the pool step.
In order to echo the last article, here I will use the identical example data, first and second steps of the MI process, and just illustrate the difference in the third step. Assume that the endpoint is the change from baseline at week 6, and given that this drug is used to reduce the primary indicator, the null hypothesis might be that the CHG in the treatment group minus the placebo group is more than -2, demonstrating that the drug efficacy is not superior to placebo.
The combined imputation with a margin of -2 as following.
MI_Superiority
Now we can find the Theta0 value is -2 rather than the usual and default 0. And the two-sided p-value is 0.4745. If we would like to obtain the one-sided p-value, an additional calculation can be done. Or just a half of a two-sided p-value is also fine, which is the same.
data super; set super; pval = (1 - probt(abs(tvalue),df));run;
Otherwise the t-statistic and p-value can also be computed by the t distribution formula, as shown below in R.
The superiority test is used as an example above, however non-inferiority test can follow the same procedure by simply altering the margin.
]]>
<p>In the previous article (<a href="https://www.bioinfo-scrounger.com/archives/mi_sas/">Understanding Multiple Imputation in SAS</a>), we talked about how to implement multiple imputation in the SAS procedure to compare the difference between the treatment and placebo groups. Let's look at how to do it in non-inferiority and superiority trials, which differ from common use.</p>