在Co-mutation and exclusion analysis in R这篇文章中提到maftools包的somaticInteractions()函数可以做Co-mutation/exclusion分析及可视化
由于结果图中展示的颜色有点问题,因此我去查看了下其源码,发现其是参考文献:Combining gene mutation with gene expression data improves outcome prediction in myelodysplastic syndromes
从这篇NC中可看出,作者的可视化功底特别厉害。。。并且意外发现作者将其可视化的源码放在的github上,地址:https://github.com/gerstung-lab/MDS-expression,粗略一看,惊为天人。。。原来用R的base作图方法竟然也能做出这么漂亮的图片。。。比如:
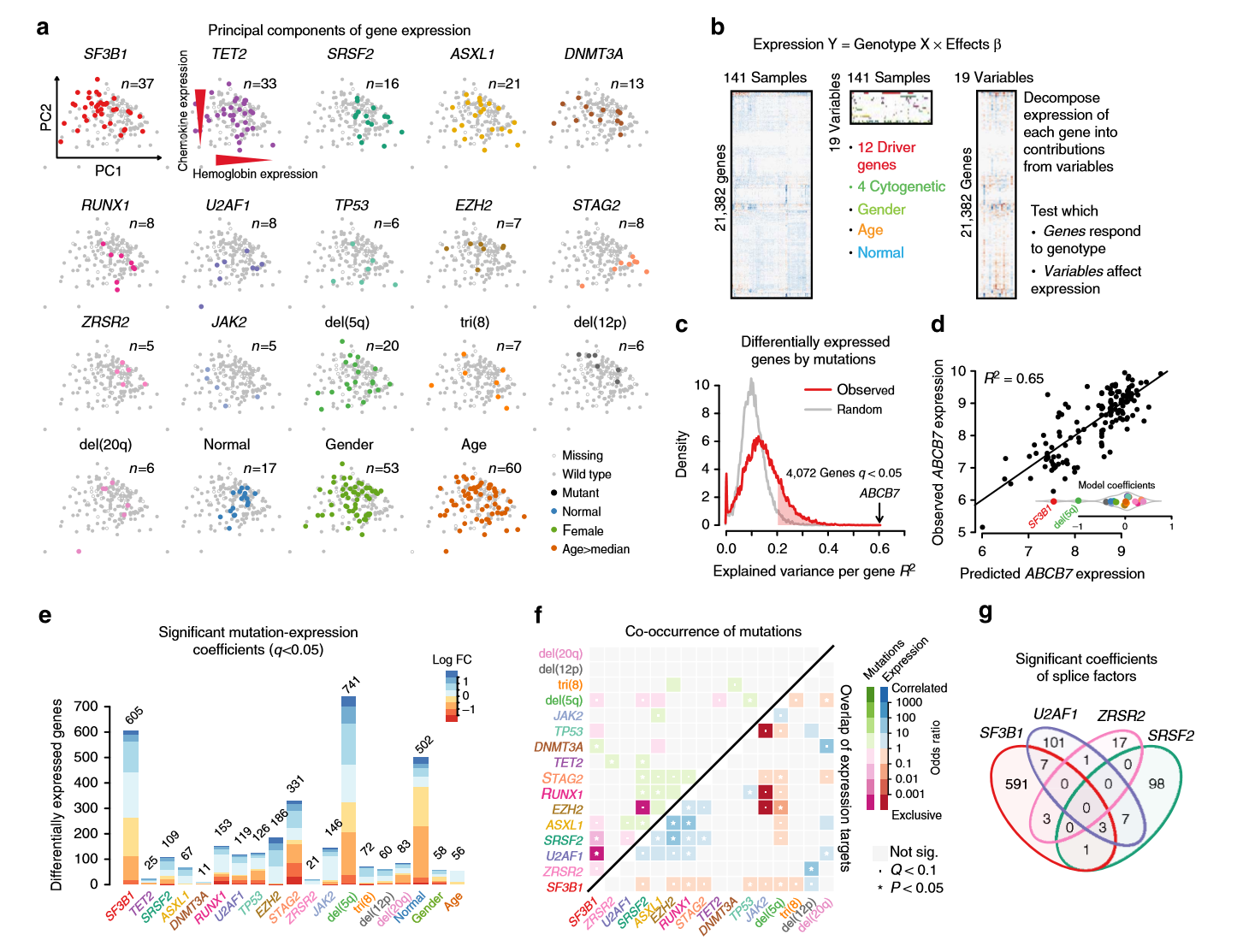
我原本是希望做出这样的可视化图片
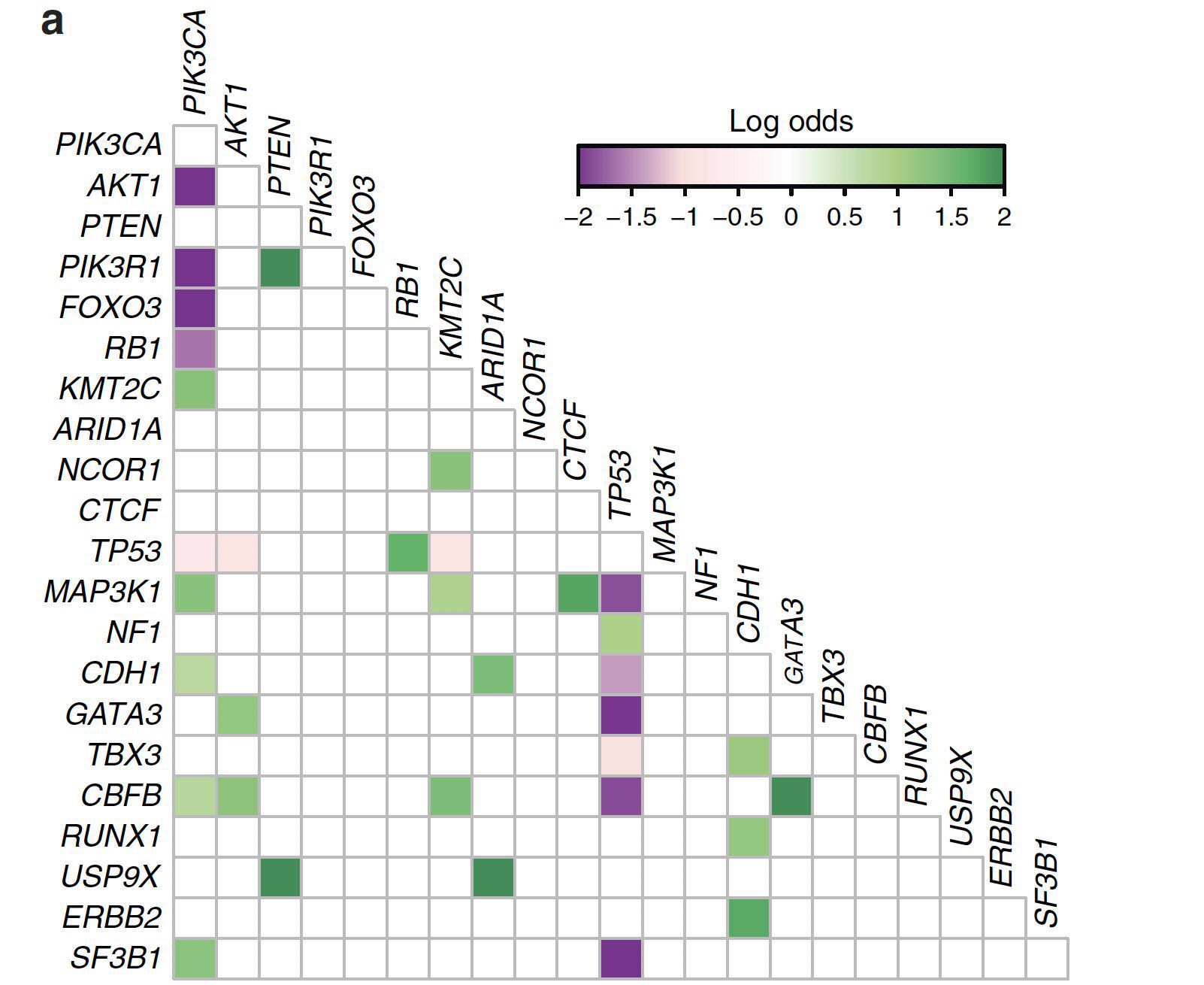
按照原来的思路:
- 修改maftools包somaticInteractions函数的源码(但是缺少点base作图的技巧,结果不太理想)
- 利用ggplot2来作图(还是没有思路)
- 利用经典相关性可视化的corrplot包(调下参修改下,可能应该可以)
由于有NC作者提供作图源码,正好可供我学习下(重点看的是第f张图所对应的代码),并对maftools包的somaticInteractions函数的图片做一定的修改(一部分也借鉴了此函数的源码),代码如下,如有兴趣可先下载我的原始数据(mutMat)
mutMat <- read.table(file = "../Desktop/mutMat.txt", sep = "\t", header = T)
interactions = sapply(1:ncol(mutMat), function(i)
sapply(1:ncol(mutMat), function(j) {
f<- try(fisher.test(mutMat[,i], mutMat[,j]), silent=TRUE)
if(class(f)=="try-error"){
NA
}else{
ifelse(f$estimate>1, -log10(f$p.val), log10(f$p.val))
}
}))
oddsRatio <- oddsGenes <- sapply(1:ncol(mutMat), function(i)
sapply(1:ncol(mutMat), function(j) {
f<- try(fisher.test(mutMat[,i], mutMat[,j]), silent=TRUE)
if(class(f)=="try-error"){
NA
}else{
f$estimate
}
}))
diag(interactions) <- NA
diag(oddsRatio) <- NA
colnames(oddsRatio) <- rownames(oddsRatio) <- colnames(interactions) <- rownames(interactions) <- colnames(mutMat)
oddsRatio[10^-abs(interactions) > 0.05] = 1
oddsRatio[oddsRatio < 0.1] = 0.1
oddsRatio[oddsRatio > 10] = 10
par(bty="n", mgp = c(2,.5,0), mar=rep(4,4)+.1, las=2, tcl=-.33)
m <- nrow(oddsRatio)
n <- ncol(oddsRatio)
r <- log10(oddsRatio)
r[lower.tri(r)] <- NA
image(x=1:n, y=1:m, r, col=brewer.pal(9,"PiYG"), breaks = c(seq(from = log10(0.1), to = 0-.Machine$double.eps, length.out = 5),
seq(from = 0, to = log10(10), length.out = 5)),
xaxt="n", yaxt="n", xlab="",ylab="", xlim=c(0, n+3), ylim=c(0, n+3))
abline(h=0:n+.5, col="white", lwd=.5)
abline(v=0:n+.5, col="white", lwd=.5)
pvalue <- c(0.05, 0.01)
text(x=n/2, y=m+1, "Genetic interactions", pos=3, cex=1.3)
w0 <- arrayInd(which(as.vector(upper.tri(r)) == TRUE), rep(m, 2))
w = arrayInd(which(10^-abs(interactions) < min(pvalue)), rep(m,2))
points(dplyr::inner_join(data.frame(w0), data.frame(w)), pch="*", col="black")
w = arrayInd(which(10^-abs(interactions) < max(pvalue)), rep(m,2))
points(dplyr::inner_join(data.frame(w0), data.frame(w)), pch=".", col="black", cex = 2)
image(y = 3:10 +6, x=rep(n,2)+c(2,2.5), z=matrix(c(1:8), nrow=1), col=brewer.pal(8,"PiYG"), add=TRUE)
image(y = 3:10 +6, x=rep(n,2)+c(2,2.5)+1, z=matrix(c(1:8), nrow=1), col=brewer.pal(8,"PiYG"), add=TRUE)
axis(side = 4, at = seq(3,11) + 5.5, tcl=-0.15, label=signif(10^seq(-1,1,length.out = 9), 2), las=1, lwd=.5, cex.axis=1)
mtext(side=4, at=12, "Odds ratio", las=3, line=3)
par(xpd=NA)
text(x=n+1.2, y=17.5, "Correlated", pos=4)
text(x=n+1.2, y=8-.7, "Exclusive", pos=4)
points(x=rep(n,2)+2, y=c(1,2.5), pch=c("*","."))
image(x=rep(n,2)+c(1.5,2.5), y=c(4,5), z=matrix(1), col=brewer.pal(3,"BrBG"), add=TRUE)
mtext(side=4, at=seq(1,4.5,length.out = 3), c(paste0("P < ", min(pvalue)),
paste0("P < ", max(pvalue)),
"Not sig."), line=0.2)结果如下:

至少看起来数据和颜色是对应上了,OR值大于1的以绿色展示,OR值小于1的以紫红色展示,不显著的以淡灰色作为背景
上述代码的一些R的base作图的技巧值得学习下。。。特别是看了NC作者的源码后,大有启发。。。
不得不说maftools真是这个神奇的工具,如果没有分析思路的话可以去其文档看看;其中的一些分析方法都是基于一些文献的,可以拓展下思路(一些方法的统计学方法并不复杂,只是在于怎么将其写出来罢了。。。);比如
OncogenicPathways函数,其是分析了TCGA已知的10个致癌信号通路,统计了maf文件中在各个通路下的基因数目以及患者人数等信息,方法比较简单,但是却很实用~
本文出自于http://www.bioinfo-scrounger.com转载请注明出处