这方法最初是在Y叔的微信软文非模式基因GO富集分析:以玉米为例+使用OrgDb中学到的,其主要讲了如何获取非模式物种的OrgDb包。而OrgDb包主要收录了其对应物种的注释信息,比如以人类的org.Hs.eg.db为例: library(org.Hs.eg.db) >columns(org.Hs.eg.db) [1] "ACCNUM" "ALIAS" "ENSEMBL" "ENSEMBLPROT" "ENSEMBLTRANS" [6] "ENTREZID" "ENZYME" "EVIDENCE" "EVIDENCEALL" "GENENAME"
[11] "GO" "GOALL" "IPI" "MAP" "OMIM"
[16] "ONTOLOGY" "ONTOLOGYALL" "PATH" "PFAM" "PMID"
[21] "PROSITE" "REFSEQ" "SYMBOL" "UCSCKG" "UNIGENE"
[26] "UNIPROT"
从上述可看出,包含了NCBI,ENSEMBL,UCSC和Uniprot等主要数据库的注释信息,还有一些GO,PFAM,OMIM等信息,相当于将这些数据库的信息通过包的形式封装了在一起(以特定格式),便于用户访问。(PS.人类研究的最为详细,所以信息量也是最大的,其他物种就没这么多注释信息了)
起初我以为要获取OrgDb包必须要去http://bioconductor.org/packages/3.5/BiocViews.html#___OrgDb才行,里面总共有19个物种的OrgDb包可供安装
注:如果不能翻墙的话,上面这个网站是进不去的,可以通过折中的方法,从https://bioconductor.org/packages/3.5/data/annotation/进,然后搜索org.,也能找到那19个物种的OrgDb包
但是通过非模式基因GO富集分析:以玉米为例+使用OrgDb这篇文章,了解到了原来有AnnotationHub包,其提供了一个非常方便的交互方式来下载不同物种(包括模式和非模式的物种)的注释资源(由Bioconductor团队统一维护更新,一般说来是跟bioconductor版本更新同步,非常方便R读取处理)
我们来看一下AnnotationHub包总共包含了哪些注释信息。首先加载包,并且会在你用户目录下创建一个隐藏文件~/.AnnotationHub,里面主要包含了annotationhub.sqlite3文件,存储了annotationhub的一些信息,从而以便下次使用时不必要再次联网下载
library(AnnotationHub)
ah <- AnnotationHub()我们可以看看AnnotationHub的注释资源的来源有哪些,这里好多我至今都还未接触过。。。
>unique(ah$dataprovider)
[1] "Ensembl"
[2] "UCSC"
[3] "RefNet"
[4] "Inparanoid8"
[5] "NHLBI"
[6] "ChEA"
[7] "Pazar"
[8] "NIH Pathway Interaction Database"
[9] "Haemcode"
[10] "BroadInstitute"
[11] "PRIDE"
[12] "Gencode"
[13] "CRIBI"
[14] "Genoscope"
[15] "MISO, VAST-TOOLS, UCSC"
[16] "UWashington"
[17] "Stanford"
[18] "dbSNP"
[19] "ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/"
[20] "URGI"我们还可以看看注释格式类型有哪些,OrgDB只是其一种数据类型,如:
unique(ah$rdataclass)其实最好的查看的方式还是通过其一个GUI命令,以交互式表格的信息的方便查看和搜索,可用于查看同一物种不同版本下的注释信息,如:
display(ah)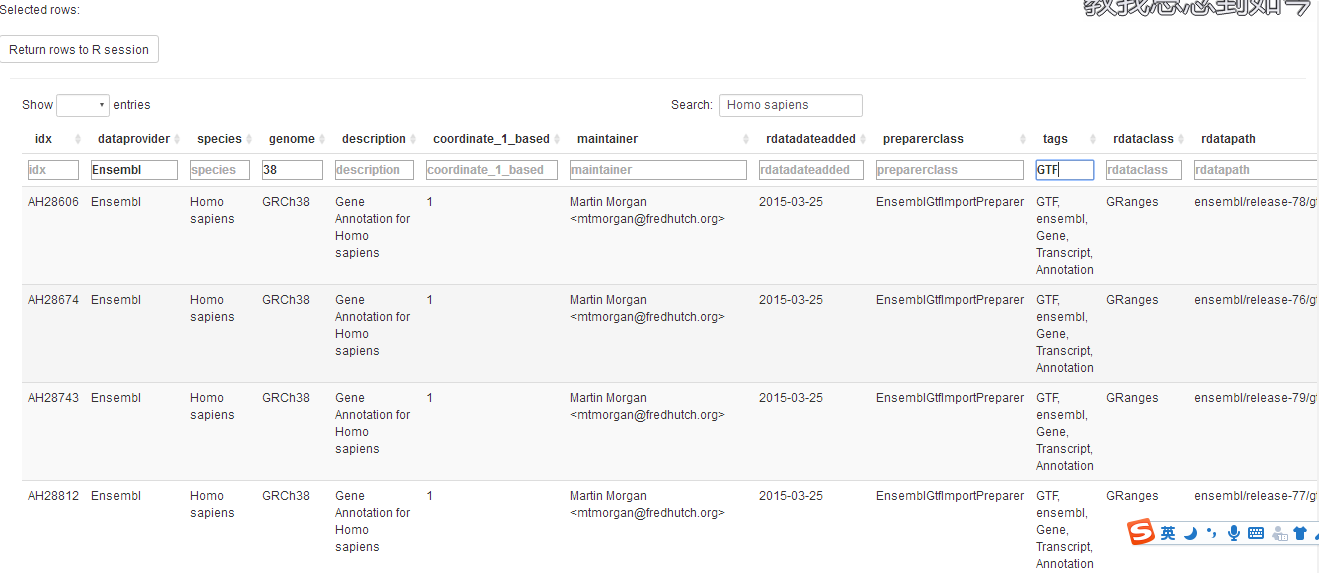
那么如何寻找对应物种的某一个注释包呢,这里以人物种的OrgDb为例,个人习惯这样:
先按物种筛一遍,然后用display函数的可视化界面慢慢找,这样比较适合不确定要找哪个注释信息,或者一些偏门的物种(也就是注释信息比较少的)
hm <- query(ah, "Homo sapiens")
display(hm)如果明确了就是要找OrgDb类型,那么可以下面的方法
org <- ah[ah$rdataclass == "OrgDb",]
hm <- query(org, "Homo sapiens")
>hm
AnnotationHub with 1 record
# snapshotDate(): 2017-10-27
# names(): AH57973
# $dataprovider: ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/
# $species: Homo sapiens
# $rdataclass: OrgDb
# $rdatadateadded: 2017-10-23
# $title: org.Hs.eg.db.sqlite
# $description: NCBI gene ID based annotations about Homo sapiens
# $taxonomyid: 9606
# $genome: NCBI genomes
# $sourcetype: NCBI/ensembl
# $sourceurl: ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/, ftp://ftp.ensembl.org/pub/...
# $sourcesize: NA
# $tags: c("NCBI", "Gene", "Annotation")
# retrieve record with 'object[["AH57973"]]'然后就是下载对应的文件org.Hs.eg.db.sqlite,可以通过下述两种途径下载,如果下载慢或者总是断了,那么换个时间段再下载吧,PS.其文件也是下载到上述的隐藏文件夹下,下次可免下载直接调用
hm_org <- ah[["AH57973"]]
hm_org <- hm[[1]]
hm_org还可以将hm_org对象保存重命名,再载入
saveDb(hm_org, file = "human.orgdb")
loadDb(file = "human.orgdb")上述是以模式物种人为例的,其实非模式物种也是这样操作,只是注释信息的多少会根据物种的注释程度高低而变化
当已经获取的OrgDb对象后,该怎么处理呢?常规的需求一般有这几种:不同数据库的ID转化,基因/转录本/蛋白的ID转化,GO注释以及其他注释,主要用到的函数有columns(),keys(),keytypes(),select()等
columns():主要看下这个OrgDb对象有哪些类型数据可以提取
keys():主要看某个类型数据下的内容又哪些(一般是ID)
keytypes():一般搭配keys()使用,如果单独使用内容其实跟columns()是一样的
select():主要讲不同数据集关联起来,并提取出数据
如:
看看hm_org包含的数据集有哪些
>columns(hm_org)
[1] "ACCNUM" "ALIAS" "ENSEMBL" "ENSEMBLPROT" "ENSEMBLTRANS"
[6] "ENTREZID" "ENZYME" "EVIDENCE" "EVIDENCEALL" "GENENAME"
[11] "GO" "GOALL" "IPI" "MAP" "OMIM"
[16] "ONTOLOGY" "ONTOLOGYALL" "PATH" "PFAM" "PMID"
[21] "PROSITE" "REFSEQ" "SYMBOL" "UCSCKG" "UNIGENE"
[26] "UNIPROT"看看REFSEQ数据集下是那些ID,不设定keytype的值则默认是ENTREZID
>head(keys(hm_org, keytype = "REFSEQ"))
[1] "NM_130786" "NP_570602" "NM_000014" "NM_001347423" "NM_001347424"
[6] "NM_001347425"还可以看能匹配Refseq id的NM_130786对应entrez id是哪个(没看错,其entrez id就是1)
>keys(hm_org, keytype = "ENTREZID", column = "REFSEQ", pattern = "NM_130786")
[1] "1"看看每个ENTREZID对应的GO ID是哪些(只要记住keytype参数永远都是对应keys参数的就行)
>head(select(hm_org, keys = keys(hm_org), columns = c("ENTREZID", "GO"), keytype = "ENTREZID"))
'select()' returned 1:many mapping between keys and columns
ENTREZID GO EVIDENCE ONTOLOGY
1 1 GO:0002576 TAS BP
2 1 GO:0003674 ND MF
3 1 GO:0005576 IDA CC
4 1 GO:0005576 TAS CC
5 1 GO:0005615 IDA CC
6 1 GO:0008150 ND BP另外还有个mapIds函数,其用法跟select类似,可以指定输出的格式,有时候可以用用
除了常见的OrgDb类型,还有TxDb类型:其主要储存了转录组的注释信息,比如一些转录本/外显子/CDS的位置,注释信息等,TxDb.Hsapiens.UCSC.hg19.knownGene是我们比较常见和使用的数据集了;还有BSgenome类型:其主要储存了序列信息,如BSgenome.Hsapiens.UCSC.hg19
上述的TxDb/BSgenome包除了可以通过常规的手段安装biocLite(),也可以用AnnotationHub()安装(如果是人的话,还是常规的安装算了,至少下载快。。。)
上述资料还参考了Genomic Annotation Resources
如果要对AnnotationHub的使用进行练习的话,上述链接下有不少Exercise可供实践哦
本文出自于http://www.bioinfo-scrounger.com转载请注明出处