我们常见的转录组表达分析一般都是将reads比对至参考基因组或者转录组上,然后在基因或者转录本水平上定量表达丰度。
转录组定量-featureCounts
在转录组定量分析时,如果采用的是alignment-based转录组定量策略,那么一般会使用的是HISAT2、STAR或者TopHat等比对软件。
接着则是对转录组进行定量,如果是基于基因水平的定量,我之前一般是采用HTSeq-count工具来获取每个基因上的count数。所谓count数,个人简单的理解为根据不同比对情况,将reads分配到各个基因上。HTSeq-coun对于多重比对的reads则是采取舍弃策略。当HTSeq-count选择默认参数(-m 默认模式),那么reads是以下图所示的union的情况进行分配的
R学习笔记 dplyr包处理数据
在生信领域,不可避免会使用R来处理一些数据,不仅因为R在生信数据分析中的广泛应用,而且也由于R其强大的数据处理以及统计分析能力。不管是NGS数据,还是芯片或者公共数据,我们都需要对其进行预处理以及相关的下游分析,这时如何提高处理及分析效率就是一个关键的技能。
起初我一般会选择一些R的base函数进行操作,但从最开始学习R开始,就听起别人说有个叫dplyr的R包是一个处理数据的利器,结果由于那时too native地觉得dplyr也就比base函数方便一点而已。直到最近使用了dplyr包后,才发现自己之前的想法是多么幼稚!现在只要是处理数据的R脚本,我会毫不犹豫的先加载个dplyr。。。。
R作图 显著性绘制工具-ggsignif
最近在逛R语言数据分析与可视化是发现了有个ggpubr包可以绘制箱线图的显著性标记,感觉蛮有用的,然后就在网上搜了下显著性标记相关资料,发现除了ggpubr包外,发现一个ggsignif包!查了下原来这个包是专门配套ggplot2来给图添加显著性标记的,所以收藏并使用了下
回顾基因表达芯片分析(数据分析)
这次以R语言处理表达芯片教程中的GSE号为例子,具体可以参看http://www.biotrainee.com/thread-566-1-1.html
回顾基因表达芯片分析(数据处理)
去年的这个时候,我参加了生信技能树Jimmy办的线上R语言处理表达芯片教程,这也是我进入生信领域,第一个学习的完整流程。那时对生信的一些基础概念还是处于模糊的状态,只是略懂了点R语言,勉强的听完了整个教程。
R作图 火山图静态/交互式可视化(ggrepel和plotly)
火山图(Volcano Plot)常用于展现差异表达的基因,横坐标常为Fold change(倍数),纵坐标为P value(P值),这里以静态和动态两种形势展示火山图,主要还是为了介绍下ggplot2的插件ggrepel和plotly包
ChIP-Seq分析小实战(三)
做完前面两部分实战总结ChIP-Seq分析小实战(一) ChIP-Seq分析小实战(二),这个实战教程也剩下了最后的peak注释以及可视化了
ChIP-Seq分析小实战(二)
在ChIP-Seq分析小实战(一)中以将数据做好了准备,接着就是按照文章中所述将ChIP-seq数据用botwie比对至NCBIM37基因组上,但是这次我还是按照教程的方法,使用bowtie2比对至mm10基因组上
ChIP-Seq分析小实战(一)
首先要先感谢生信技能树Jimmy分享ChIP-seq基础入门教程,让我有机会以及动力去好好了解ChIP-seq,教程的提纲以这篇2013年发表在Cell Reports的文章RYBP and Cbx7 define specific biological functions of polycomb complexes in mouse embryonic stem cells为例,并给出了这篇文章ChIP-seq分析的每个步骤,可供我们实践。
Perl学习笔记 引用和匿名(数组/散列/子例程)
引用
Perl的标量变量保存单个值;数组保存一个有序的标量变量;散列保存一个无序的标量集合作为值
个人觉得,正是因为有引用,将标量、数组和散列很好的结合在一起,才在处理复杂的数据结构中游刃有余,引用可以分别数组引用和散列引用,还有个是子例程引用
Perl学习笔记 数组/散列(哈希)
数组
数组中的每个元素都是单独的标量变量,并且是有序的,我们可以通过数组索引对其赋值
@array = ()定义空数组,$#array表示数组最后一次元素的索引值,$array[0]表示数组第一个元素以及$array[-1]表示数组最后一个元素等
怎么理解数组是有序这个意思呢,比如我想将有三个元素的数组赋值于一个只有两个元素的数组,那么后面的数组的第三个元素将自动被忽略,如下:
($one, $two) = ("aaa", "bbb", "ccc");或者这么理解:
@array[2,3] = ("a", "bb");rMATS差异可变剪切分析
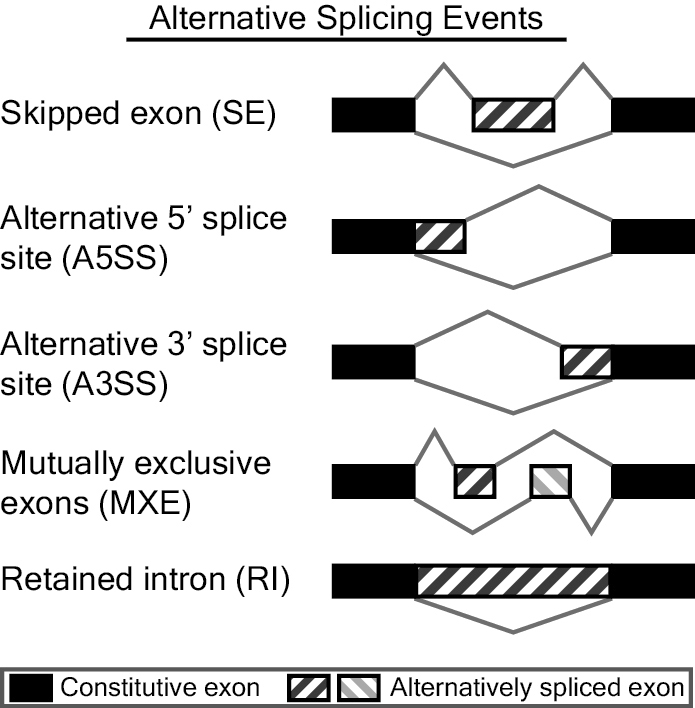
rMATS是一款对RNA-Seq数据进行差异可变剪切分析的软件。其通过rMATS统计模型对不同样本(有生物学重复的)进行可变剪切事件的表达定量,然后以likelihood-ratio test计算P value来表示两组样品在IncLevel(Inclusion Level)水平上的差异(从公式上来看,IncLevel跟PSI的定义也是类似的),lncLevel并利用Benjamini Hochberg算法对p value进行校正得FDR值。rMATS可识别的可变剪切事件有5种,分别是skipped exon (SE)外显子跳跃,alternative 5' splice site (A5SS)第一个外显子可变剪切,alternative 3' splice site (A3SS)最后一个外显子可变剪切,mutually exclusive exons (MXE)外显子选择性跳跃和 retained intron (RI)内含子滞留,展现形式如下图(来自官网http://rnaseq-mats.sourceforge.net/index.html)
Perl学习笔记 grep|map|sort操作符
《入门书籍》: Perl语言入门 ,Perl进阶
grep操作符
grep操作符可以通过表达式or块形式,将列表中的每个元素按照顺序依次取出,然后根据一定形式对其求值,最后将满足条件的元素赋值于某个列表。
在不了解grep操作符之前,如果想对列表中的元素进行处理提取符合要求的元素时,通常只能用foreach循环列表,但这样是不效率的做法。
Htseq Count To Fpkm
我们通过HTseq-count对hisat2比对后的bam文件进行计数后,会得到每个基因上比对上的reads数,也就是通常所说的count数。接着如果需要比较不同样本同个基因上的表达丰度情况,则需要对count数进行标准化,因为落在一个基因区域内的read counts数目一般可以认为取决于length of the gene(基因长度)和sequencing depth(测序深度),所以引申出两种标准化方法:RPKM和FPKM。前者是以每个reads作为一个单位,在单端测序中应用较多;而后者是以fragment作为一个单位,主要应用在双端测序后的分析。
SAM格式的学习
Sam格式在NGS中是一个不可或缺的格式,所以我们必须对其有一定了解。网上有很多文章对其有讲解,我当初也是看了那些文章入门的,当然最后只懂了每列是干嘛用的,但是具体每个标识代表什么含义却一知半解。虽然这不影响后续的学习以及相关的分析,但是对于这个一个这么重要的格式来说,这是不够的。
比如我们都知道SAM格式分为头部分和比对部分,那么头部分每个符号代表什么意思呢
TCGA 临床数据的提取
TCGA数据库除了有各种癌症的大样本数据外,还有很完善的临床数据,这点是其他数据库所缺少的,因此我们需要会从TCGA数据库中提取对应癌症的临床数据,然后利用这些数据来进行后续的分析(比如想知道某个基因的表达对病人的预后是否有影响)
STRINGDB包的简单使用
String是一个很好的蛋白互作网络数据库,其不仅包含了直接物理作用的互作关系,还包含了蛋白之间以间接作用的互作关系。除了有实验证据支持的数据外,还有整合其他数据库中的互作数据以及利用生物信息学预测获得的互作数据。
TCGA lncRNA的提取
众所周知lncRNA属于RNA中的非编码RNA,在转录调控中扮演者重要角色。并且现在听说lncRNA的研究也很火热,使用TCGA的数据对lncRNA的研究也是非常常见的需求。而我们如果想对TCGA的lncRNA进行定量则必须从TCGA RNA-Seq的表达量数据中提取出lncRNA的那部分数据。
TCGA ID转化的小插曲
在下载TCGA数据后,我们会发现新版的TCGA表达数据有6万个ENSG(ENSEMBL ID),听说旧版时是用gene symbol的,而且我也觉得有时gene symbol更能说明问题。如果用过Firehose下载的TCGA数据的话,会发现其是用gene id和gene symbol共同来表示表达数据的。如果是使用生信人的那款工具的话,其也是提供了ID转化功能,即把ENSG转化为gene symbol。