FCM(Fuzzy C-Means)算法是一个模糊聚类算法,是对早期硬K-means聚类的一种改性,克服了硬聚类的非此即彼的分类缺点。
FCM属于软聚类,它允许一个数据点可以属于多个类,FCM的价值函数跟K-means非常相似,但其引入了隶属度的概念,使得每个数据点用值在[0,1]的隶属度来确定其属于各个组的程度,其结果是每个数据点对聚类中心的隶属程度(membership),取值在0-1之间,并进行归一化使得总和等于1;因此FCM的思想是使得被划分到同一组下的数据点之间相似性最大,而不同组之间的相似度最小 FCM的C跟K-means的K是相同的概念,是指聚类的数目,Fuzzy是指一个事件的发生程度(模糊相似关系),因此FCM算法需要两个参数:聚类数目C和模糊参数m。一般来说,C要远远小于聚类样本(蛋白/基因)数目,并保证至少大于1;而m(fuzzifier值)是控制算法柔性的参数,用于定义整个数据集的模糊度,一般默认是2,接近于1的话会导致结果接近于K-means聚类,因此不同的C和m会导致聚类结果的不同
FCM聚类算法主要涉及隶属度(隶属矩阵u)、聚类中心v(距离)以及模糊参数m等参数,即在满足隶属度约束条件下,通过迭代计算价值函数的最小值;求约束条件的求极值问题,使用拉格朗日乘子法来构建拉格朗日函数,具体公式推导可看FCM(Fuzzy C-Means)模糊C聚类,算法大致步骤如下:
- 初始化隶属矩阵(满足价值函数的约束条件),计算聚类中心;或者初始化聚类中心
- 迭代计算价值函数,当其小于某个极小值或者前后两次的差值小于某个极小值,则停止
- 更新隶属矩阵,确定聚类结果
由于FCM算法依赖于初始化参数,所以需要使用一些其他方法获取一个比较好的聚类中心以及隶属矩阵;或者使用多个初始聚类中心,进行多次计算取最优的的那个(即nstart参数的作用)
对于模糊参数m的选择,一般都默认取2,因为一般取2能达到较好的效果。但是A simple and fast method to determine the parameters for fuzzy c–means cluster analysis文献对于基因表达谱数据以及质谱(iTRAQ)数据做了研究表明,可以通过数据集的样本数目以及纬度(基因/蛋白数目)来判断最佳的fuzzifier值
一般对于C的选择也可以通过不同的validation indices来评价,比如有:Partition Entropy (PE)、Partition Coefficient (PC)、Modified Partition Coefficient (MPC)以及Xie-Beni index(XB)等等
简单的FCM算法R语言实现代码如下(先随机取3个点作为初始聚类中心v0,计算欧几里得距离d,确定隶属矩阵u,然后迭代至价值函数j的变化差异小于极小值,更新隶属矩阵u):
FCM <- function(x, k, m, iter.max = 1000, con.val = 1e-09){
x <- as.matrix(x)
set.seed(12345)
v0 <- x[sample(nrow(x), 3),]
n <- nrow(x)
d <- matrix(0, n, k)
for (j in 1:k) {
d[, j] = sqrt(rowSums(sweep(x, 2, v0[j, ], "-")^2))
}
u <- matrix(NA, nrow = n, ncol = k)
for (j in 1:k){
for (i in 1:n){
if (any(d[i,] == 0)){
u[i, ] <- rep(1/k, k)
}else{
u[i, j] <- 1/(sum((d[i, j]/d[i, ])^(2/(m - 1))))
}
}
}
v <- t(u^m) %*% x/colSums(u^m)
j <- sum(d^2 * (u^m))
iter <- 0
j_best <- Inf
while((iter < iter.max) && (abs(j - j_best) > con.val)){
j_best <- j
for (j in 1:k) {
d[, j] = sqrt(rowSums(sweep(x, 2, v[j, ], "-")^2))
}
for (j in 1:k){
for (i in 1:n){
if (any(d[i,] == 0)){
u[i, ] <- rep(1/k, k)
}else{
u[i, j] <- 1/(sum((d[i, j]/d[i, ])^(2/(m - 1))))
}
}
}
v <- t(u^m) %*% x/colSums(u^m)
j <- sum(d^2 * (u^m))
iter = iter + 1
}
result <- list()
result$u <- u
result$v <- v
return(result)
}上面的FCM代码只是写写,熟悉下算法,一般实际中会使用现有的R包中的函数来计算;FCM聚类算法可用R包有:e1071包的cmeans函数,ppclust包的fcm函数;前者出的较早,被其他包引用的较多,而后者17年刚出,整合了多个聚类的算法,功能齐全以及使用较为方便
表达谱FCM聚类的可视化方式有:Mfuzz包,或者用R语言绘图;前者Mfuzz包调用了e1071包的cmeans函数,所以如果用Mfuzz包的话,必须使用其整合的cmeans函来做FCM聚类;如果自行绘图的话,可以使用ppclust包来做FCM聚类
ppclust包的使用说明可以参考Partitioning Cluster Analysis Using Fuzzy C-Means
测试数据
data(iris)
x=iris[,-5]其可以通过inaparc包的kmpp函数来计算初始聚类中心或者imembrand函数来计算初始隶属矩阵
v0 <- inaparc::kmpp(x, k=3)$v
res.fcm <- fcm(x, centers=v0)
u0 <- inaparc::imembrand(nrow(x), k=3)$u
res.fcm <- fcm(x, centers=3, memberships=u0)或者使用现有的初始化算法来计算初始隶属矩阵u以及初始聚类中心v,ppclust包的fcm函数也是调用了inaparc包
res.fcm <- fcm(x, centers=3, alginitv="hartiganwong", alginitu="imembrand")如果不想使用欧式距离,可以通过dmetric参数更换距离计算方法
res.fcm <- fcm(x, centers=3, dmetric="correlation")如果想计算多个初始化中心,则用nstart参数,并用fixcent和fixmemb来分别保持初始聚类中心和隶属矩阵不变(两者不能同时存在),然后以最小的价值函数值对应的结果作为最优解
res.fcm <- fcm(x, centers=3, nstart=5, fixmemb=TRUE)最后用plotcluster函数对两两特征值进行绘图展示,或者使用factoextra包的fviz_cluster函数来可视化展示(需要先将fcm函数的结果格式转化为kmeans的,然后再绘图)
res.fcm <- fcm(x, centers=3, alginitv="hartiganwong", alginitu="imembrand", nstart = 5, fixmemb=TRUE)
res.fcm2 <- ppclust2(res.fcm, "kmeans")
factoextra::fviz_cluster(res.fcm2, data = x,
ellipse.type = "convex",
palette = "jco",
show.legend.text = FALSE)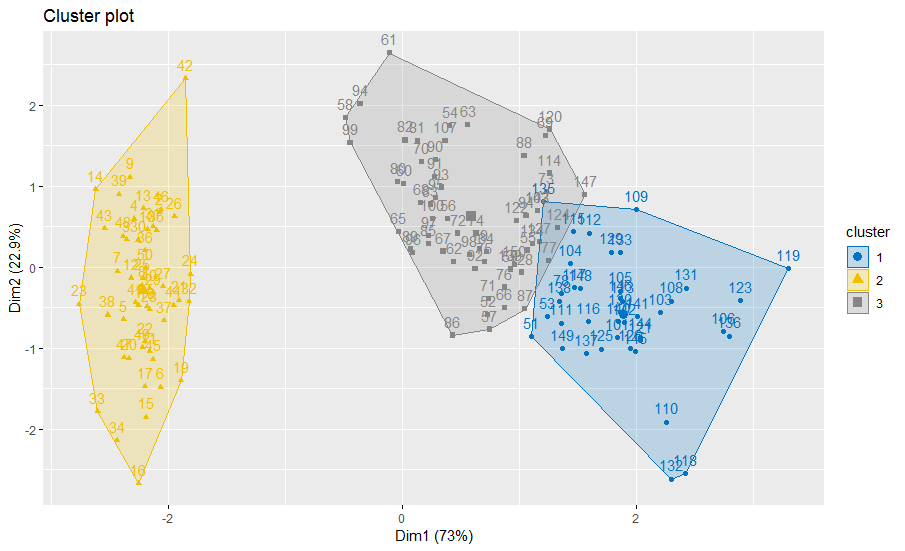
plotcluster(res.fcm, cp=1, trans=TRUE)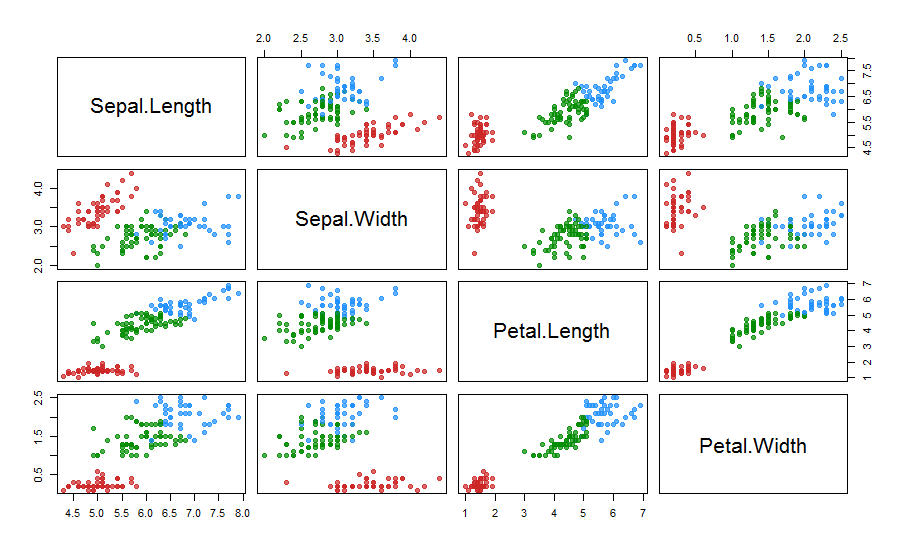
可以跟原始数据集的分类情况比较下,真实分类情况如下:
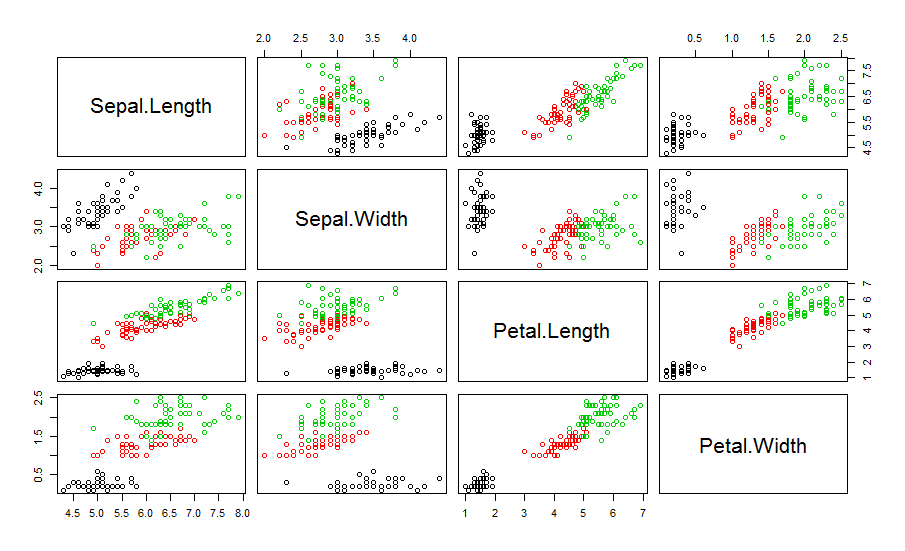
为了验证C值的选择,先将fcm函数的结果格式转化为fclust格式,然后用PE或者PC函数来看看可能最优的C值
res.fcm2 <- ppclust2(res.fcm, "fclust")
idxpe <- PE(res.fcm2$U)
idxpc <- PC(res.fcm2$U)对于基因/蛋白等表达谱数据,可以用Mfuzz包对FCM聚类结果进行可视化处理,其调用了e1071包的cmeans函数做FCM分析,然后将聚类结果以热图曲线形式展示,以一个测试数据为例:
library(Mfuzz)
data(yeast)
eset <- new("ExpressionSet", exprs = rawdata)
eset <- standardise(eset)
m <- mestimate(eset)
cl <- mfuzz(eset, c = 5, m = m)
mfuzz.plot(eset, cl, mfrow=c(3,2), new.window= FALSE, time.labels = c("CK", "12h", "24h", "48h"))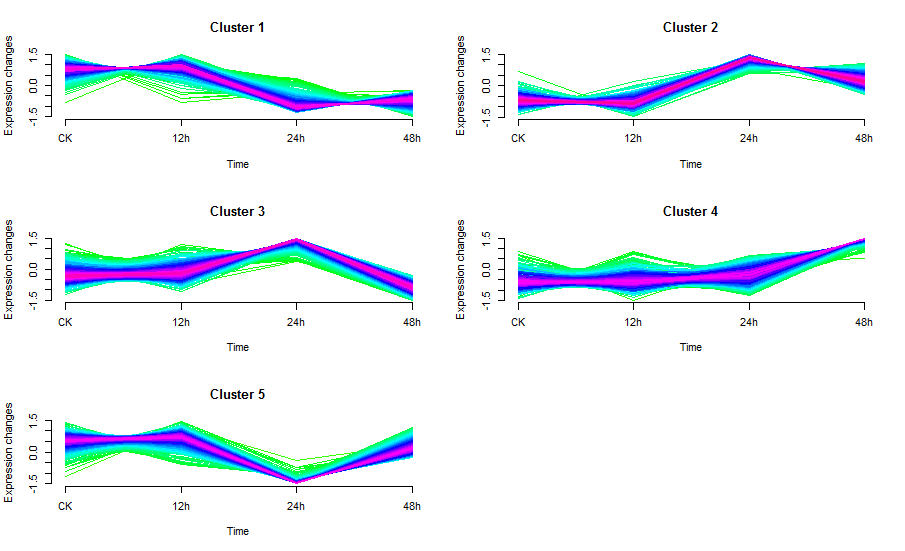
图中线条颜色越接近红色的说明是这些基因/蛋白是这个cluster中的core,他们有着较高的membership值;这个包的作者默认设定了0.7作为core的阈值,即membership大于0.7作为这个cluster的core
如果想展示其中单个cluster的结果,则如下:
mat <- matrix(1:2, ncol=2, nrow=1, byrow=TRUE)
l <- layout(mat, width=c(6,1), heights = c(1,5))
mfuzz.plot2(eset, cl, mfrow = NA,
# colo="fancy",
col.sub = "blue",
col = "blue",
cex.main = 2,
single = 2, x11 = FALSE,
time.labels = c("CK", "12h", "24h", "48h"))
mfuzzColorBar(main="Membership", cex.main=1)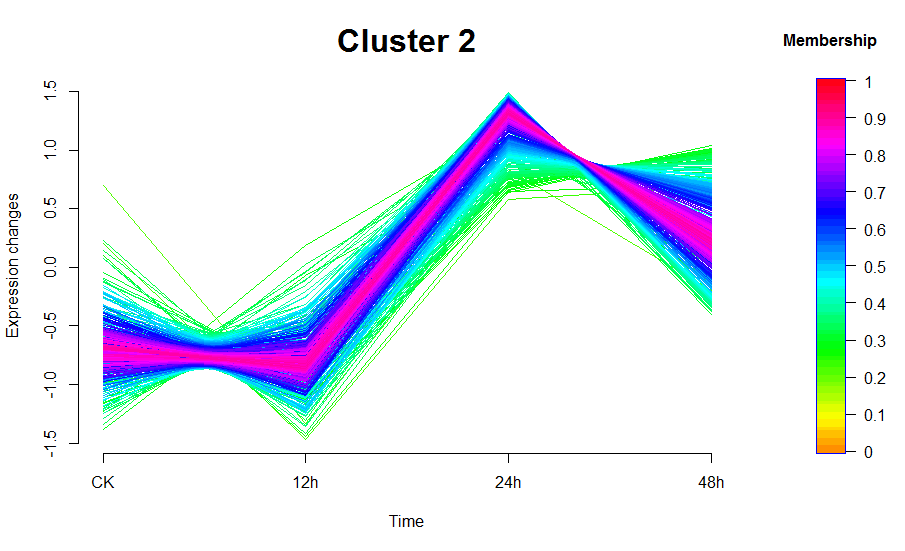
如果想确定哪个C值比较合适,则先通过对不同的C值用e1071包的cmeans函数计算一遍,然后用fclustIndex函数计算每个指标,根据不同指标的性质来判断最佳的C值
cl <- cmeans(x = rawdata, centers = 5, iter.max = 1000, method = "cmeans")
resultindexes <- fclustIndex(cl, rawdata, index="all")
resultindexes["xb"]参考资料:
FCM聚类算法介绍
模糊聚类(FCM)介绍及R语言实现
Partitioning Cluster Analysis Using Fuzzy C-Means
http://bioconductor.org/packages/release/bioc/vignettes/Mfuzz/inst/doc/Mfuzz.pdf
本文出自于http://www.bioinfo-scrounger.com转载请注明出处