说到富集,富集是将基因根据一些先验的知识(也就是常见的注释)进行分类的过程。我们一般会想到最常见的是GO/KEGG富集,其思路是先筛选差异基因,然后确定这些差异基因的GO/KEGG注释,然后通过超几何分布计算出哪些通路富集到了,通常会选择一个阈值来卡一下,比如p值和FDR等。因此这会涉及到人为的阈值选择,具有一定的主观性,而且只能用于差异较大的基因,所以结果可能有一定的局限性。 根据上述情况,有了GSEA(Gene Set Enrichment Analysis),其思路是发表于2005年的Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles,主要是要有两个概念:预先定义的基因集S(基于先验知识的基因注释信息)和待测基因集L(一般是表达矩阵);然后GSEA目的就是为了判断S基因集中的基因是随机分布于L(排序后的数据集),还是聚集分布在L的顶部或者底部(这也就是富集)。如果待测基因集中的某些基因显著富集在L的顶部或者底部,这说明这些基因的表达(因为其是根据表达谱数据)对你定义的分组(预先分组)的差异有显著影响(一致性),从而找到我们关注的基因集;在富集分析的理论中,GSEA可以认为是第二代,即Functional Class Scoring (FCS) Approaches
GSEA的使用
这里不详细说算法,具体可看GSEA的文章,因为我也是一知半解。。。
下载地址http://software.broadinstitute.org/gsea/downloads.jsp,PS.会验证下你的邮箱,先注册下
第一次使用的话,而且数据不大的话,建议使用javaGSEA Desktop Application,Java的图形化界面,使用较为友好点,根据你的数据大小,选择不同内存的版本[1-8G不等],PS.记得看看系统的Java版本,2G内存开始的GSEA版本需要的是64位的Java 1.8版
打开后的界面如下:

数据准备
因为GSEA分析一般只作用于人物种的,所以我准备以TCGA的BRCA的mRNA数据作为测试数据,正好也试下UCSC xena 浏览器才是最简单的TCGA数据下载途径这个方法来下载TCGA数据(数据还蛮新的,2017年的)
其还提供了ID/Gene Mapping的文件(整理好的),正好可以拿来用,因为虽然GSEA有EnsemblID转化的chip文件,但是感觉有些数据有点问题(可能是由于Ensembl的版本一直在更新的缘故),HUGO gene symbol最好
然后用R处理下,将癌组织和对应的癌旁组织的数据分别提取出来分别作为两组的表达矩阵(gct文件)以及或者分组文件(cls文件)
library(dplyr)
library(stringr)
data <- read.table(file = "TCGA-BRCA.htseq_fpkm.tsv", sep = "\t", header = T, stringsAsFactors = F, check.names = F)
data_df <- data[, -1]
normal_df <- data_df[, str_sub(names(data_df), 14, 15) %in% 11:19]
length(names(normal_df))
tumor_df <- data_df[, grepl(paste(str_sub(names(normal_df), 1, 12), collapse = "|"), names(data_df)) & !names(data_df) %in% names(normal_df)]
length(names(tumor_df))
idmapping <- read.table(file = "gencode.v22.annotation.gene.probeMap", sep = "\t", header = T, stringsAsFactors = F)
geneid <- data.frame(id = data$Ensembl_ID, stringsAsFactors = F)
geneid2symbol <- left_join(geneid, idmapping, by = "id")
all_df <- cbind(Name = geneid2symbol$gene, DESCRIPTION = "na", tumor_df, normal_df)
group <- c(rep("Tumor", 118), rep("Normal", 113))
group <- paste(group, collapse = " ")
group <- c(paste(c(118+113, 2, 1), collapse = " "), "# Tumor Normal", group)
write.table(file = "BRCA_fpkm.txt", all_df, sep = "\t", col.names = T, row.names = F, quote = F)
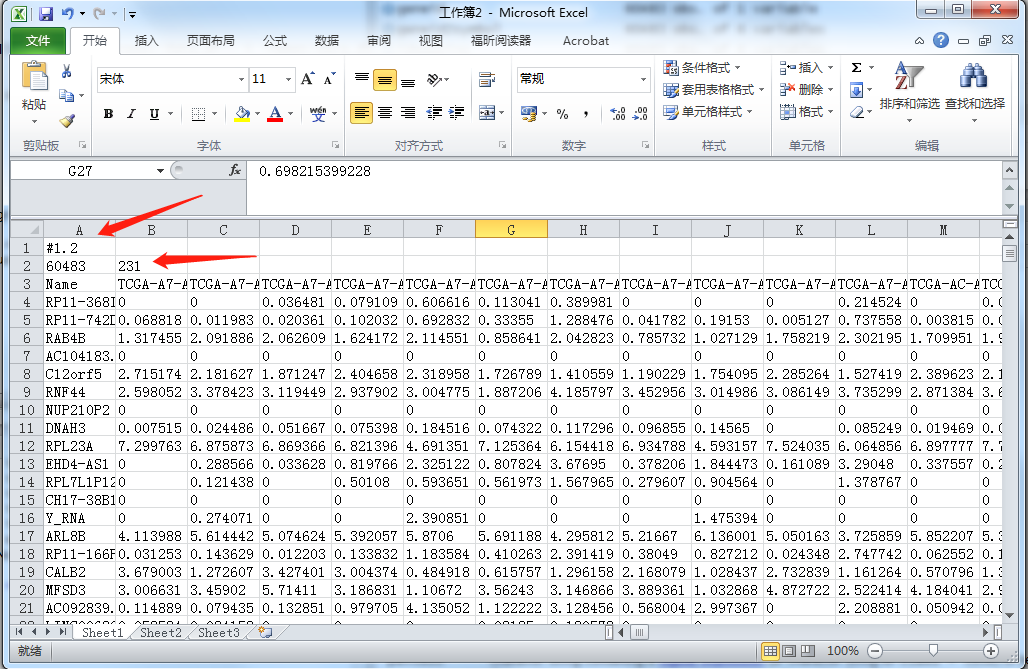
write.table(file = "group.cls", group, col.names = F, row.names = F, quote = F)从上述代码,我获得118个癌组织样本和对应的113个癌旁样本的表达谱数据,并且将Ensembl ID均转化为了Gene symbol(避免之后用GSEA时,再用chip做ID转化);然后可以直接将txt文件作为输入,也可以将BRCA_fpkm.txt文件做一些处理变成GSEA标准的gct文件,如下图所示:分别加上前2行内容,第一行照抄就行,第二行则是geneid数目和样本数目
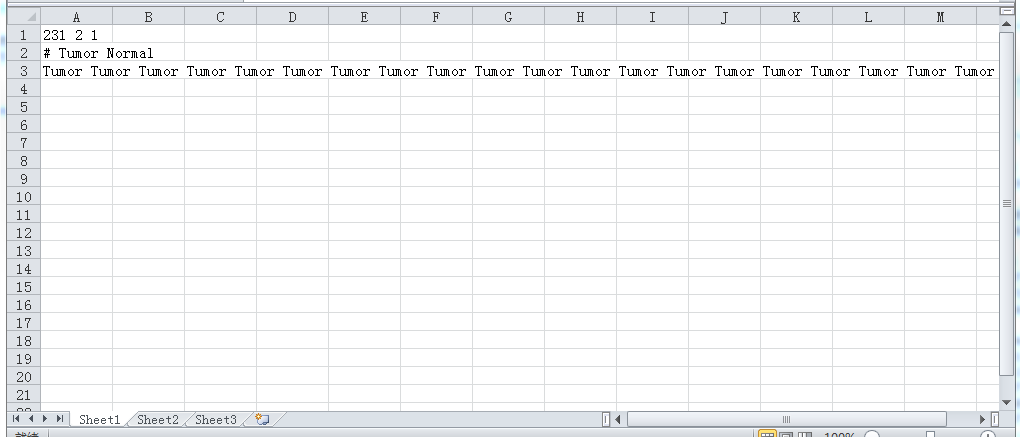
接着是Phenotype labels文件(上述代码直接出了),即cls文件,格式如下图所示:第一行231代表样本数目,2代表分2组,空格间隔,1照抄;第二行井号注释说明分组信息;第三行为每个样本对应的组名,空格分隔
上述文件的详细格式可参照网站:http://software.broadinstitute.org/cancer/software/gsea/wiki/index.php/Data_formats
如果网络不佳的话,接下来最好将Gene sets file(也就是GSEA软件上需要输入的Gene sets database),作者将gene sets都储存在Signature Database (MSigDb)中,去官网下载即可http://software.broadinstitute.org/gsea/downloads.jsp,比如下载个c2.cp.kegg.v6.1.symbols.gmt文件作为Gene sets database
如果数据是芯片数据或者需要GSEA的chip文件做ID转化的话,则也可以先将chip文件下载下来,FTP地址:ftp://ftp.broadinstitute.org/pub/gsea/annotations
软件使用
因为是windows桌面式软件,使用就比较简单了。首先将BRCA_fpkm.txt和group.cls文件导入,如果网速不好的话(比如我自己),那也把上述下载下来的c2.cp.kegg.v6.1.symbols.gmt文件也导入
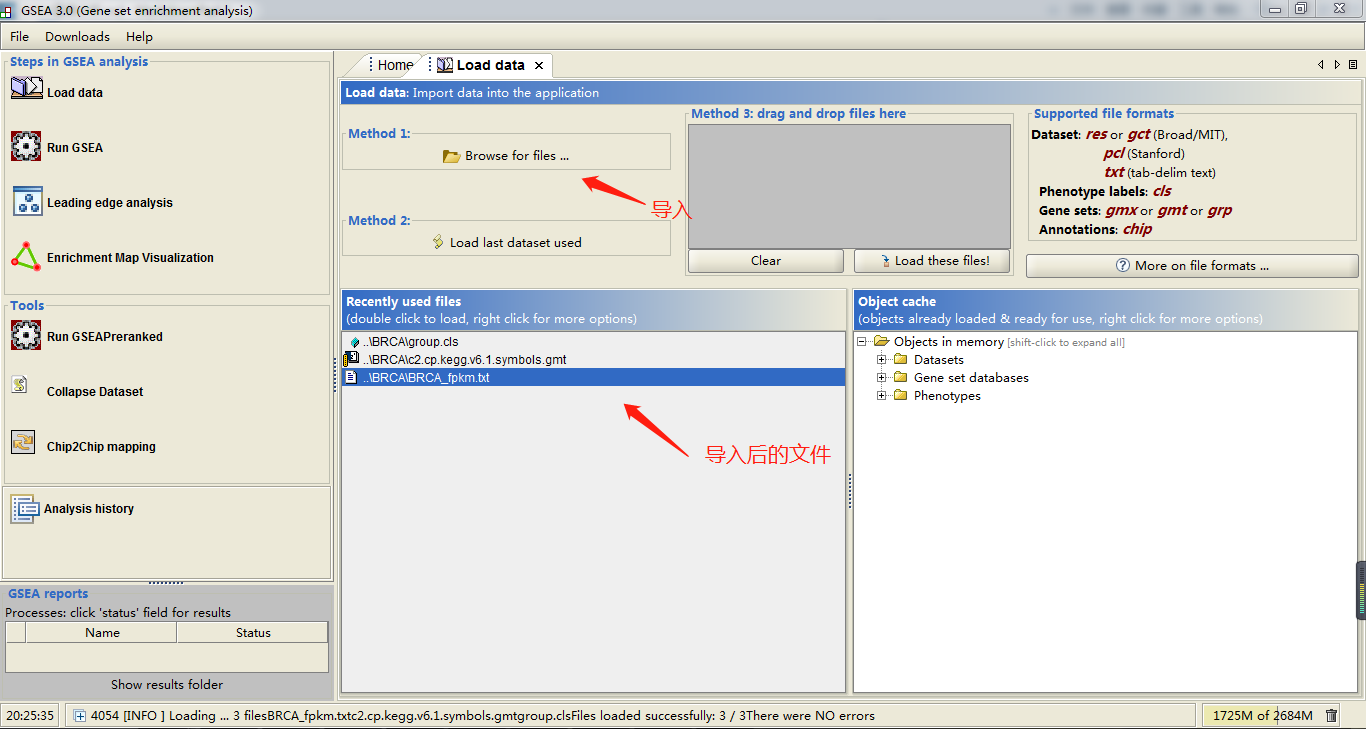
接下来点击RUN GSEA,就是几个指定参数的选择了,如下图所示:

- Expression dataset:输入的表达矩阵
- Gene sets database:gene sets文件,这里是
c2.cp.kegg.v6.1.symbols.gmt文件;PS.这个文件也可以自己创建哦- Number of permutations:置换检验的次数
- Phenotype labels:选择比较组,如果你输入的文件就只有2个组别的话,这个就很方便选一个就行了;如果你输入的有三个组别及以上的话,则这里就要跟你的需要选择两个组别的比较组,而且GSEA也会根据你的组别信息去表达矩阵中提取相对应的数据
- Collapse dataset to gene symbols: 如果你已经ID转化为HUGO gene symbol,那么这里选FALSE,否则选择TRUE
- Permutation type:选择置换的类型,是random phenotype还是random gene sets,一般每组样本数目大于7个时,建议选择phenotype,否则选择gene sets(这句话一直没在官网上找到。。。似乎在文章里?)
- Chip platform:早些时候主要都是芯片数据,那时必须要将芯片id转化为HUGO gene symbol,所以这个参数一直叫做chip(我猜的。。。),其实就是对ID进行注释,即ID转化,选择你ID对应的chip文件即可,如果已自行转化了ID的话,则这里空着就行(记得Collapse dataset to gene symbols选择否)
除了Required field参数外,下面还有Basic fields和Advanced fields,具体参见官网吧(注:或者鼠标悬浮在对应参数名称上,有简单的参数介绍哦)
最后点击RUN,等待左下角的Running变成Success,然后点击Success即可查看完整的结果,也可以点击Show results folder,GSEA将所有结果都放在一个文件夹中了!!!
分析结果
来看下文章里最常见的GSEA的结果图片,如下图所示:
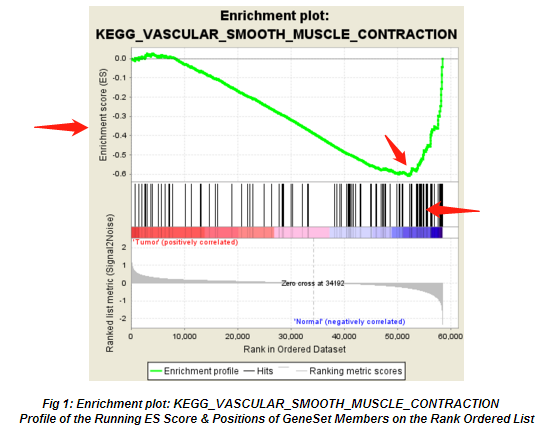
从图上,我们一般关注ES值,峰出现在前端还是后端(ES值大于0在前端,小于0在后端)以及Leading-edge subset(即对富集贡献最大的部分,领头亚集);在ES图中出现领头亚集的形状,表明这个功能基因集在某处理条件下具有更显著的生物学意义;对于分析结果中,我们一般认为|NES|>1,NOM p-val<0.05,FDR q-val<0.25的通路下的基因集合是有意义的
除了上述的结果外,GSEA还提供了Running the Leading Edge Analysis等操作,也可以看看
GSEA的结果解读我也不是太熟悉,还是得多看看文献中的解释说明啦
多于多个样本的批处理,GSEA也有服务器版本,通过命令行即可操作,适合批处理操作;其还提供了R脚本可供使用(但官网上说似乎并一定可行,需要自己调整?),反正我也正准备都试试看。。。
参考资料:
功能数据库专题-GSEA
GSEA富集分析 - 界面操作
http://software.broadinstitute.org/gsea/doc/desktop_tutorial.jsp
http://software.broadinstitute.org/gsea/doc/GSEAUserGuideFrame.html
GSEA使用(初级)
本文出自于http://www.bioinfo-scrounger.com转载请注明出处