本来应该这是一个很正常的学习过程,之前总结了一篇博文Bioconductor的质谱蛋白组学数据分析,对蛋白组学定量那块比较感兴趣,正好看到一个R包-MSstats,其可用来对DDA,SRM和DIA的结果进行蛋白差异分析,这R包发表于2014年,那时来说还是很不错的(还在不断更新维护),并且其还支持Maxquant查库结果文件作为输入(主要我有些此类测试文件),非常有兴趣的想尝试下看看结果,然后就入坑了。。。 #### 初步使用
从其官网http://msstats.org/可看出,其现在的功能还是非常全面的,当然我只暂时用到其一小部分功能
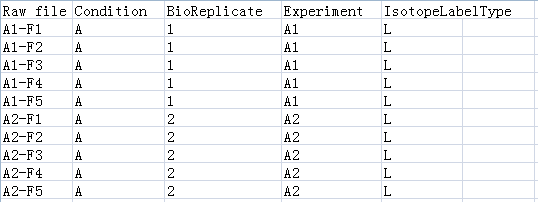
正常流程一般是从Biocondutor上其R包的使用说明看起,然后拿自己的测试数据走一遍流程。但是!!从第一个函数MaxQtoMSstatsFormat(用于将Maxquant查库结果文件转化为其标准输入格式)起就遇到了一个槛,由于其需要一个annotation文件(感觉类似于甲基化芯片分析的Champ包那个注释文件),但其官网文档并没有给出完整的格式,翻了一些资料并看了这函数的源码才整理出一份正确的格式(我的是Label-free分级测试文件),如下:
本以为后续应该可以比较正常的进行下去了,结果在第二个对输入数据进行处理的函数dataProcess就开始不停的报错,我本以为是我测试数据的问题,结果换了好几批数据后才觉得,可能是我的数据跟其认为的maxquant的正常输出结果不一致,因此决定从其源码开始找寻报错的原因,结果从其代码中发现了几处'BUG'(至少对我的测试数据来说)
其实对R包这种开源的软件来说,如果遇到报错信息,首先想到的是查阅官网网站或文档寻找答案;再者去网上寻找是否有跟自己遇到相同问题的人,并且看看是否有对应的解决办法;最后如果还是不行的话,那么就看R包对应函数的源码吧(至少能解决不少问题)。其实有些R包并不复杂,而且看源码的过程也是一种学习的过程,等以后自己写R包的时候也能用上一些技巧嘛
下面则是我看了MSstats包的几个重要函数后的随笔,记录了个人理解下的其运行的原理(主要其发表的文章中并未提起原理部分,只是粗略讲了软件的功能以及结果展现形式,所以还是得从代码入手),但有些地方还是不太理解
dataProcess函数
下面这段摘抄自其官网文档,主要讲了这个函数做了哪些事情:
Data pre-processing and quality control of MS runs of the original raw data into quantitative data for model fitting and group comparison. Log transformation is automatically applied and additional variables are created in columns for model fitting and group comparison process.
- Summary report: feature, sample, missingness (warning messages)
- Logarithm transformation with base 2 or 10
- Normalization: bias of MS run
- Equalize median normalization
- Quantile normalization
- normalization using global standard proteins
- Equalize median normalization
- Feature selection: all features, top3 features, or topN features
- Model-based run quantification
- Tukey’s median polish or linear model
- Label-based or label-free
- Censored or random missing values
- Imputation by accelerated failure model or not
- Tukey’s median polish or linear model
对于FRACTION
一个样本,循环,第一个run1开始,对应去重后的肽段,统计每个样本在不同run下的有abundance的肽段个数,将同个样本中最大数目肽段的run对应的FACTION赋予对应的run1的肽段,循环至最后一个run,然后停止。
其原理我认为是这样的:在MS分级中,会将样本等量分成N份,对应的就是N个run,从而将不同特性的离子区分开;那么在某个run里鉴定到肽段数目一般相比其他run来说应该是最大的
代码中有个问题,其只循环了样本1,而其他样本则不管了,这里会导致一些肽段万一没有在样本1出现,那么其对应的FRACTION则是NA,会导致代码报错;但如果再循环每个样本的话,后面的样本会影响前面样本的结果,个人觉得应该每个样本各自确定自己RUN对应的FRACTION即可。最终我觉得这步骤主要还是为了确定FRACTION而已
我这里有个疑问,为啥不根据结果的RUN的那列数据来确定FRACTION呢,至少命名上还是可以看出不同RUN到底是属于哪个FRACTION的。PS.如果做了5次分级,6个样本,那么其实了跑了30次LC/MS,那么有30个RUN,但是FRACTION还只是5次哦
不同FRACTION下肽段丰度的选择
对于同一肽段在不同FRACTION下有不同的丰度(abundance),那么该如何确定哪个abundance应该赋予这个肽段,这个也是我之前一直想知道的,作者则是做了以下处理:
- 如果肽段只在一个FRACTION中被测到(也就是有丰度值),则保留
- 如果肽段在多个FRACTION被测到,那么在某一FRACTION下检测到丰度数目最多的(也就是被测到次数最多的)那些肽段的丰度值被保留,其他的丰度值则去除
- 接着去除那些肽段在所有RUN中总共只被测到一次的,可能是杂音吧?
- 如果在多个FRACTION下出现的肽段次数是一样的,那么计算这些FRACTION下,该段丰度平均值的大小,去除掉丰度平均值较小的肽段的丰度值
- 以上的去除并不是删掉肽段,而是将对应FRACTION下的肽段的丰度/强度值变为NA
Normalization
经过上述步骤过滤完后,则肽段只有在某一FRACTION下才有丰度值,其实主要是针对分级的Label-free数据而言的(PS. 真心觉得Label-free还是分级的好,不分级鉴定数目还是少了点);而言也做了Logarithm transformation,接下来则是标准化了,作者主要给了以下几个选项:
constant normalization (equalize medians)
肽段丰度值-RUN下丰度中位数+FRACTION下丰度中位数
quantile normalization
preprocessCore包的
normalize.quantiles函数,原理参照分位数标准化global standards
就是以某肽段(数目可以多个)作为内标进行标准化
还可以将多个标准化的结果进行合并,也就是不同的run用不同的标准化方法,最后合并
除了标准化外,作者还用了Tukey's median polish(TMP)算法确定一个cutoff,根据这个阈值过滤掉低丰度值的肽段(即将这些肽段的丰度值变为NA),涉及的默认参数是summaryMethod,censoredInt和MBimpute
此外,作者对于那些Protein Group只有一个肽段的(也就是说这个Protein Group只是由一个肽段定性出来的,可信度比较低。。。),并没有舍去,只是给了提醒,让用户考虑是否要舍去
Model-based run quantification
最后这步是这个函数比较重要的一个统计分析,其以一个Protein Group为一个单位,依次进行分析(也就是一个Protein Group一次分析),主要做了以下处理:
- 根据
summaryMethod参数的模型不同,其结果有略有差异,主要分TMP和linear两大类 - 接着对一个Protein Group内的肽段进行过滤,如果一个肽段其只被测到一次,则过滤
- 过滤掉没有丰度值的肽段
- 如果设定了
remove50missing参数,则会过滤掉那些缺失值超过50%的run - 接着用了survival::survreg函数对丰度值(ABUNDANCE)做了个拟合模型(大概是这个意思),从而对丰度值进行校正
最终的结果为一个processedquant列表用于后续分析
dataProcessPlots函数
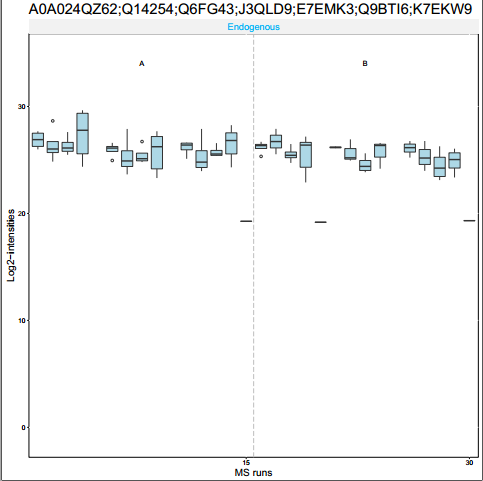
这个函数主要是用于可视化的,展示一个或者N个Protein Group下的肽段丰度分布,主要有箱线图和折线图等,用一下就知道了,主要输入对象是上面的processedquant
groupComparison函数
这个函数则比较重要了,Finding differentially abundant proteins across conditions,也就是差异蛋白分析,也是我一直比较想了解的分析过程。虽然使用很简单,但是既然上面几个步骤的源码都看了,也不差这一个函数了,顺便也看了一遍,归纳下主要是以下几个步骤:
使用的话,可以分为两步,先建立比较组矩阵(类似NGS那些差异分析软件的分组矩阵),然后就用groupComparison进行分析了,如下:
comparison <- matrix(c(-1,1),nrow=1)
row.names(comparison) <- "B-A"
comparison_result <- groupComparison(contrast.matrix = comparison, data = processedquant)
head(comparison_result$ComparisonResult)接下来我想通过源码了解作者是如何在Protein层面上进行差异分析(结果我关键的算法步骤还是没看懂,统计方面实在太差,以后再补上了),其他步骤大概是这样的:
- 先根据你设定的比较组矩阵依次进行差异分析
- 每次比较组的差异分析是以Protein Group为单位进行的
- 一个Protein Group下面有多个肽段,并且可能都在两个样本中被测到,那么正常出FC和p-value,如果只有在其中一个样本中被测到,则都为NA
- 主要有两种算法,默认是linear model,这是根据上个函数dataProcessPlots得到的结果中的SummaryMethod是否为logOfSum决定的,但是默认都是TMP,则对应的就是linear model,个人觉得跟limma中的lm算法可能有点关系;还有种算法则是logsum t-test,这个就比较好理解了,其实就是做了T-test,而且FC值也很容易看懂,是两个样本的平均丰度值的差值,为什么是差值呢,因为在之前处理中就已经做了log2转化了
- 剩下的SE,Tvaue,DF则是一些统计量
- 总体上可看出其是根据Protein Group下不同样本的肽段的丰度值来进行差异分析的,我怎么看感觉其是将每个肽段的丰度值看成了一个'取样'来处理了(主要是看logsum t-test算法的,linear model没这么看懂,所以也就不确定了);不像一些蛋白组定量中的T-test检验,是针对同一个Protein Group对应不同样本的丰度值(这就是基于蛋白水平的丰度值了)
Summary
折腾了一周,每天断断续续看了其代码,虽然代码比较长,但是很多部分是一些数据判定检查过程(可跳过),所以代码思路非常严谨,从其代码中也学到了不少小技巧,也算一个额外的收获。并且还了解了作者对于蛋白组定量差异分析的一些思路,可能不是最优的,但也是一个参考。虽然这个R包对我的数据来说无法正常使用(因为必须先修改其函数中的部分代码才行),但理解其思路才是最主要的
本文出自于http://www.bioinfo-scrounger.com转载请注明出处