这里并不是全面地介绍如何对蛋白质组学进行分析,而是利用这篇文献Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research,从一款用于蛋白组学下游分析的软件的角度,来写写常规的分析流程。从个人的角度来看,现在蛋白组学数据的上游分析因受仪器等原因的限制,分析方法没有较大的进步,分析流程也都整合在一些商业化软件中了,开源的软件较少。而下游分析则比较简单,还处于类似测序的芯片时代(意思就是分析内容几乎跟芯片分析一样,就那几种,唯一区别:蛋白组是基于肽段/蛋白表达矩阵。。。) 这篇文章是发表于2018年Methods in Molecular Biology丛书中第七章,主要介绍了如何使用Perseus这款软件来对蛋白组学数据进行下游分析。我没使用过这软件,所以主要结合自己对蛋白组数据的一些了解,借助这篇文章来梳理下分析流程。蛋白组学数据个人理解可以分为两部分:图谱数据和表达矩阵数据,前者是质谱的下机数据;然后一般会用一些熟知的软件进行分析转化,将峰信号转化为肽段/蛋白的表达矩阵数据。在这篇文章中,测试数据是基于Labelfree技术以及MaxQuant软件的结果文件,样本分组为正常样本、肿瘤样本以及转移组织样本
分析流程
以这篇文章的思路,分析步骤主要分为以下几步:
Preprocessing
拿到MaxQuant的结果文件,首先需要先去除污染蛋白(这类蛋白是在查库时匹配到了reverse database or contaminants),接着做log2转化(这个作者也说了,可做可不做);如果有需求,还可以对空值进行补充,由于质谱仪检测的限制,一些蛋白的在某样本中的表达量为能达到检测的最低线,从而会导致空值的产生。
Filtering
这里的过滤并不是选择差异表达的蛋白,而是基于表达量,选择一定的数值进行整体上的过滤(比如根据缺失值在样本中的数量对蛋白进行过滤,或者根据表达量的阈值进行过滤等等)
Exploratory Analysis
主要是可视化展示数据,并看看是否有离群样本等,主要通过样本间/组间相关性分析,以聚类热图或者PCA散点图展示,这些可视化形式跟NGS数据一样,都可以用R进行处理
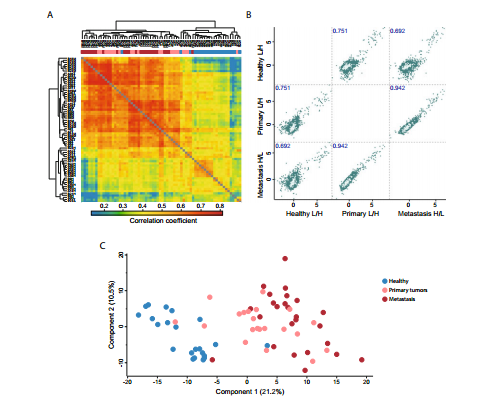
Proteomics_pca Normalization
这里的标准化并不是NGS中一些软件根据一定的分布模型进行的标准化,而是以一些常用的标准化方法对数据整体上做个归一化而已,如Z-score标准化
Annotations
注释是所有组学数据中最常见的分析步骤,特别是在转录组中;而蛋白组数据的注释跟转录组有略微的不同,因为其ID有时是Uniprot ID,而不是Gene ID,但整体上的思路是一样的。个人觉得,如果是Uniprot ID的话,直接通过Uniprot数据库进行注释就行了,如通过Unirpot的API接口。这款软件则是通过一个mainAnnot.homo_sapiens.txt.gz文件选项来实现的,相当于将所有人的Uniprot ID所对应的注释信息都放在这个文件中,好处是不需要联网去网站上读取数据了,缺点则是这个注释文件更新则是个大问题
Expression Analysis
差异表达分析我认为是蛋白组学下游分析中最主要的步骤,特别是在大样本的时候,有一个靠谱差异分析方法是比较重要的,这篇文章主要提了两种T-test检验和ANOVA/Post-hoc tests。上述检验的前提都是蛋白组的表达值符合正态分布,那么为什么不使用芯片那套的limma分析软件呢,或者一些NGS的差异分析软件(有一些文章提到过,也准备好好看一看相关方面),总觉得在临床大样本的时候,单纯的T检验的检测效用并不够
Clustering and Profile Plots
这步主要做的是聚类分析和表达谱的可视化,一般是在上一步Expression Analysis后进行,如下图所示,B图主要是针对A图中一部分蛋白进行的表达趋势的分析,C图则是在B图中各个分类的蛋白基础上进行的功能富集分析

Proteomics_cluster Functional Analysis
这步也是在Expression Analysis基础上,一般是GO/KEGG注释后再进行富集分析,跟其他组学一样,没啥区别
Summary
这篇文章最后的Notes我觉得写的蛮实用的。其中对于T检验这步,如果样本的不是配对样本,那么T检验的前提必须是方差齐性,其实有些蛋白组数据是未达到这个前提的;作者还介绍了用Tukey’s honestly significant difference (THSD)来做post-hoc test;还提到了一点,如果差异蛋白过少或者背景蛋白过多都会对富集结果造成偏差,所以一般蛋白组学的富集分析不会选用某物种的全部蛋白作为富集分析的背景蛋白,而是只以鉴定到的蛋白为背景蛋白,因此如果差异蛋白的过少的话,你的富集结果也会很少,可能几乎都没富集到。
本文出自于http://www.bioinfo-scrounger.com转载请注明出处