本文开头先记录一篇文章李航博士的《浅谈我对机器学习的理解》
K-means聚类是一种无监督学习,主要用于对未标记的数据进行分组,这里的K代表分组的个数。K-means算法会迭代分配每个数据点到K个分组中,从而使得数据点基于特征相似性进行聚集
K-means一般认为的计算方法如下:
- 初始化簇的中心
- 遍历整个数据集,将每个数据点分配到离它最近的中心所在的簇中
- 以每个簇的数据点的均值中心点代替之前的中心,继续迭代直至收敛 我这有个基因表达矩阵DEG_file.txt,4个样品,1724个基因,如:
data <- read.table(file = "DEG_file.txt", sep = "\t", header = T, row.names = 1, stringsAsFactors = F)
dim(data)
#[1] 1724 5先对这个表达矩阵做些预处理,如挑选出变异系数较大的前1000个基因的表达矩阵,主要为了减少一些表达量变化并不大的基因对后续分析的干扰
data$cv <- apply(data, 1, function(x){
sd(x)/mean(x)*100
})
data_df <- data[order(data$cv, decreasing = T)[1:1000], 1:4]
dim(data_df)
#[1] 1000 4我希望能从矩阵中找出在一些样品中特异性高表达的基因,其实一般的层次聚类热图也能粗略看出来
data_scale <- as.data.frame(t(apply(data_df,1,scale))) ##Z-score标准化
names(data_scale) <- names(data_df)
library(ComplexHeatmap)
library(circlize)
Heatmap(data_scale,name = "heatmap",
column_names_side = "bottom",
col = colorRamp2(c(-2, 0, 2), c("green", "white", "red")),
cluster_columns = FALSE,
row_dend_side = "left",
show_row_names = FALSE,
clustering_method_rows = "complete",
)结果图如下,大概能看出基因至少能分成2-3组:
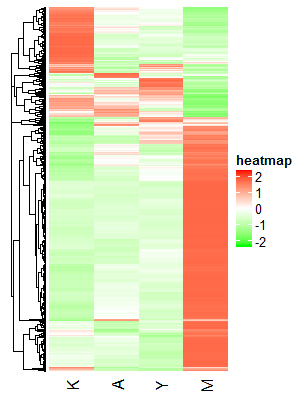
如果我们想用K-means算法根据基因表达量对其分组的话,我们首先需要知道最佳的K值,R语言:K-means 找到最佳的K值这篇文章中主要介绍了6种求最佳K值的方法,如: >1. 寻找SSE的拐点 >2. 通过分割算法来估算类的中心 >3. Calinsky标准:诊断多少集群适应数据 >4. 贝叶斯信息准则: 基于层次聚类的期望最大化 >5. 基于Affinity propagation (AP)聚类 >6. 基于Gap Statistic
还有一种可能使用较多的是基于轮廓系数,所以我准备综合使用:寻找SSE的拐点,基于Gap Statistic以及轮廓系数,来确定最佳的K值
寻找SSE的拐点
SSE(sum of the squared error),即所有点到其所属簇中心的距离的平方和即误差的平方和
wss <- (nrow(data_scale)-1)*sum(apply(data_scale,2,var)) for (i in 2:8) wss[i] <- sum(kmeans(data_scale,centers=i)$withinss) plot(1:8, wss, type="b", xlab="Number of Clusters",ylab="Within groups sum of squares")
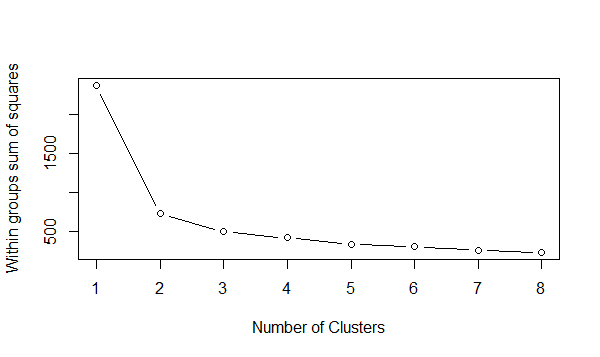
从上图可以看出,拐点(在这个拐点之前,代价函数值下降得很快,在该点之后,代价函数值下降得很慢)最佳是在2,3也可以基于Gap Statistic
library(cluster) gap_stat <- clusGap(data_scale, FUN = kmeans, nstart = 25, K.max = 8, B = 300, verbose = interactive()) plot(1:8, wss, type="b", xlab="Number of Clusters",ylab="Within groups sum of squares")
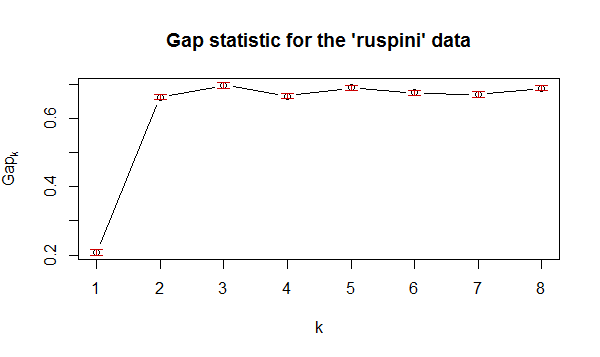
从上图可看出,最佳的K值也是在2-3。。。基于轮廓系数
轮廓系数(silhouette coefficient)方法结合了凝聚度和分离度,可以以此来判断聚类的优良性,其值在-1到+1之间取值,值越大表示聚类效果越好
K <- 2:8 round <- 30# 每次迭代30次,避免局部最优 rst <- sapply(K,function(i){ print(paste("K=",i)) mean(sapply(1:round,function(r){ print(paste("Round",r)) result <- kmeans(data_scale, i) stats <- cluster.stats(dist(data_scale), result$cluster) stats$avg.silwidth })) }) plot(K,rst,type='l',main='轮廓系数与K的关系', ylab='轮廓系数')
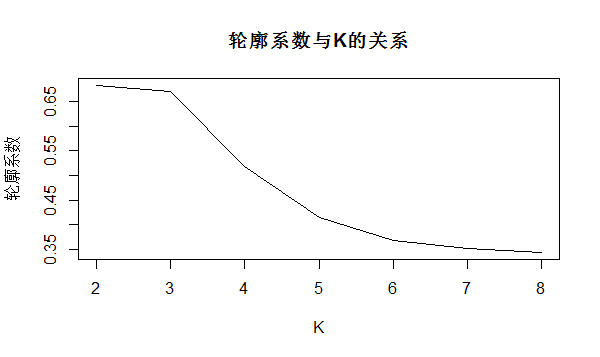
从上图可看出,最佳的K值也是在2-3。。。综上所述,我暂时认为最佳的K值为3(PS.因为我有4个样本),那么我在重新做个K-means聚类的热图
p <- Heatmap(data_scale,name = "heatmap",
km = 3,
column_names_side = "bottom",
col = colorRamp2(c(-2, 0, 2), c("green", "white", "red")),
cluster_columns = FALSE,
row_dend_side = "left",
show_row_names = FALSE
# km_title = "%i"
)
p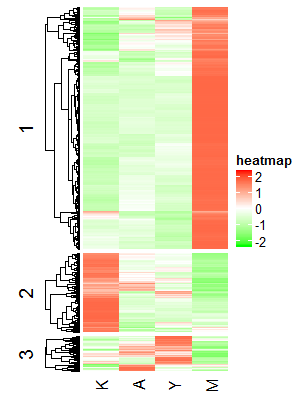
然后看看每个分类下基因在各个样本中的表达趋势
# Combined data
cls <- plyr::ldply (row_order(p), data.frame)
names(cls) <- c("id", "rowid")
cls <- dplyr::arrange(cls, rowid)
data_scale$cluster <- cls$id
res <- plyr::ddply(data_scale, "cluster", function(x){
colMeans(x[,1:4])
})
data_scale$group <- row.names(data_scale)
res$group <- "mean"
data_scale2 <- dplyr::bind_rows(data_scale, res)
df <- data.table::melt(data_scale2, id = 5:6, variable.name = "sample")
df$col <- ifelse(df$group == "mean", "red", "grey")
df$cluster <- as.numeric(df$cluster)
# Generate plot
library(ggplot2)
library(grid)
grid.newpage()
pushViewport(viewport(layout = grid.layout(1,3)))
vplayout = function(x,y){viewport(layout.pos.row = x,layout.pos.col = y)}
for (i in 1:3){
p <- ggplot(dplyr::filter(df, cluster == i), aes(x = sample, y = value, group = group, col = col)) +
geom_line() +
theme_light() +
scale_colour_manual(values = c("grey", "red")) +
guides(col = FALSE)
print(p,vp = vplayout(1,i))
}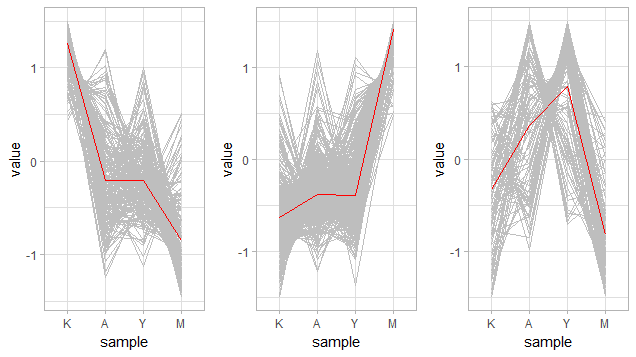
感觉第3张图分的并不是很清楚诶,可能K值还不够大?还是展示形式的问题?还是K-means算法不适合这个测试数据?暂时先这样了。。。
参考资料:
R语言学习笔记之聚类分析
R语言:K-means 找到最佳的K值
k-means 聚类k 选择(轮廓系数)
kmeans聚类理论篇K的选择(轮廓系数)
本文出自于http://www.bioinfo-scrounger.com转载请注明出处