一次偶然的搜索中发现biocondutor有个甲基化芯片的分析流程,刚好可以学习下,写的真的很棒。 Bioconductor的DNA methylation workflow可以在http://www.bioconductor.org/help/workflows/methylationArrayAnalysis/中查看,教程开头先对DNA甲基化芯片及其原理做了简单的介绍,包括一些常见的术语:比如β value和M value,后面就正式进入DNA甲基化的分析方法的讲解了。 #### 下载测试数据
测试数据是放在methylationArrayAnalysis中的,安装官网的方法安装下即可(PS.如果安装时卡住,可以用Rstudio的Tools来安装)
source("http://bioconductor.org/workflows.R")
workflowInstall("methylationArrayAnalysis")安装后,比如我R包是默认安装的R文件夹中的,所以我的数据路径是E:/R-3.4.1/library/methylationArrayAnalysis/extdata/,那么可以列出该目录下有哪些文件看看
list.files("E:/R-3.4.1/library/methylationArrayAnalysis/extdata/", recursive = TRUE)测试数据总共有两套数据(GSE49667和GSE51180),前者总共包括10个样本,作为主要的测试数据;后者则只用了其一个样本,作为测试数据中的异常样本。样本分组信息:从3个体(M28, M29, M30)中取4种不同的T-cell types(naive, rTreg, act_naive, act_rTreg),act_naive、act_rTreg是指对应经anti-CD3、anti-CD28 antibodies处理的naive、rTreg,以上分组信息也可以看数据目录下的SampleSheet.csv文件
作者提了最近几年常见的DNA甲基化芯片分析的软件,如minfi,methylumi,wateRmelon,missMethyl,ChAMP和charm,最后还提到了用limma来做差异甲基化位点分析
读入数据
先按照教程加载其所会用到的R包
library(limma)
library(minfi)
library(IlluminaHumanMethylation450kanno.ilmn12.hg19)
library(IlluminaHumanMethylation450kmanifest)
library(RColorBrewer)
library(missMethyl)
library(matrixStats)
library(minfiData)
library(Gviz)
library(DMRcate)
library(stringr)其中minfi,IlluminaHumanMethylation450kanno.ilmn12.hg19,IlluminaHumanMethylation450kmanifest,missMethyl,DMRcate是DNA甲基化分析专用R包,RColorBrewer,Gviz是用于可视化的R包,stringr,matrixStats是用于一些数据处理的,limma则是用于差异甲基化分析的
用minfi包的read.metharray.sheet函数读入IDAT格式的数据,其中SampleSheet.csv文件是必须的,不然最后数据集中会缺少样本信息;其产生的targets向量中包含了样本信息外,还有IDAT格式数据的所在路径,方便read.metharray.exp函数读入数据
targets <- read.metharray.sheet("E:/R-3.4.1/library/methylationArrayAnalysis/extdata", pattern="SampleSheet.csv")
rgSet <- read.metharray.exp(targets=targets)质控
以detection p-values评估每样本中的每个CpG位点的质量,当p-values > 0.01时则说明该探针信号数据质量较低,需要去除
detP <- detectionP(rgSet)将平均detection p-values > 0.05的样本认为是低质量的样本;从结果中可以看出最后一个样本的平均detection p-values远远大于0.05,因此从rgSet中去除(从rgSet的结构中看出,其数据分布是按照每列一个样本来的)
keep <- colMeans(detP) < 0.05
keep
rgSet <- rgSet[,keep]标准化
现在有很多用于DNA甲基化芯片数据的标准化方法,其中一些方法已经整合在minfi包中了;现在为止没有一种标准化方法被认为是最好的方法,但是一些标准化方法在某些情况下是比较好的选择,作者总结了如下几点:
- 对于用于cancer/normal or vastly different tissue types的差异比较的话,可以用
preprocessFunnorm函数(Functional normalization)- 对于不需要进行差异比较的单样本数据,如单个组织样本,那么可以用
preprocessQuantile函数- 更多的标准化方法的比较,作者推荐看A Data-Driven Approach to Guide the Choice of an Appropriate Normalization Method
除了上述提的2个标准化方法外,minfi包还支持preprocessIllumina(类似于Illumina的Genome Studio的标准化方法),preprocessSWAN(Subset-quantile within array normalisation)以及preprocessNoob(normal-exponential out-of-band)
作者比较了这些标准化方法后,发现对于这次的测试数据,这几种标准化结果都大体上相似的,所以作者最终选择了用preprocessQuantile,标准化为rgSet从RGChannelSet对象变成了mSetSq的GenomicRatioSet对象,表示从探针的信号强度转化到了beta value/M value,并且已mapping to Genome
mSetSq <- preprocessQuantile(rgSet)我们也可以不做任何标准化处理,只为获得GenomicRatioSet对象用于下游分析
mSetRaw <- preprocessRaw(rgSet)然后用minfi包内置的densityPlot函数,看看标准化前后的beta value的密度曲线
targets <- targets[keep,] ##在样本信息中也去掉异常样本
par(mfrow=c(1,2))
densityPlot(rgSet, sampGroups=targets$Sample_Group,main="Raw", legend=FALSE)
legend("top", legend = levels(factor(targets$Sample_Group)), text.col=brewer.pal(8,"Dark2"), cex = 0.6)
densityPlot(getBeta(mSetSq), sampGroups=targets$Sample_Group, main="Normalized", legend=FALSE)
legend("top", legend = levels(factor(targets$Sample_Group)), text.col=brewer.pal(8,"Dark2"), cex = 0.6)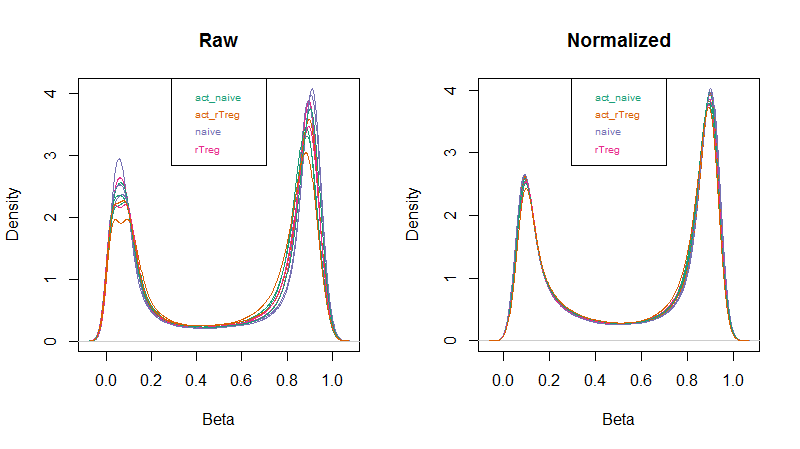
Data exploration
作者建议可以先做个主成分分析,如Multi-dimensional scaling (MDS) plots来看下样本分布情况,可以根据情况自行选择target向量中的分组,如看下targets$Sample_Group下的MDS plot,PS.这里是用M value来作图的
pal <- brewer.pal(8,"Dark2")
plotMDS(getM(mSetSq), top=1000, gene.selection="common", col=pal[factor(targets$Sample_Group)])
legend("top", legend=levels(factor(targets$Sample_Group)), text.col=pal, bg="white", cex=0.7)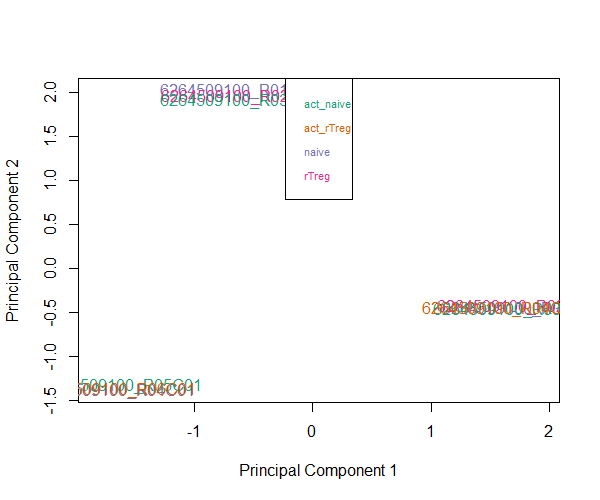
数据过滤
一些质量较差的探针需要在下游差异分析之前去除,之前的质控作者是用detection p-value去除了异常样本,在这里则需要去除在一些样本中质量较差的探针
因为前面质控时获得的
detP中的探针顺序已经和mSetSq中的探针不一致了,所以先将前者的顺序调整为和后者一致detP <- detP[match(featureNames(mSetSq),rownames(detP)),]作者以remove any probes that have failed in one or more samples为标准去除不满足detection p-value阈值(0.01)的探针,也就是说,一个探针只要detection p-value > 0.01就去除
keep <- rowSums(detP < 0.01) == ncol(mSetSq) table(keep) mSetSqFlt <- mSetSq[keep,]过滤掉X,Y染色体上的探针(一般数据都要做这一步,只要还需进行下游差异分析的话),但这个测试数据都是男性样本,所以就没必要去除了,但作者还是给出了该怎么去除在X,Y染色体上探针的代码,原理就是根据
ann450k = getAnnotation(IlluminaHumanMethylation450kanno.ilmn12.hg19)这个注释文件的数据集,里面标注了那些探针出现在哪个染色体上的信息keep <- !(featureNames(mSetSqFlt) %in% ann450k$Name[ann450k$chr %in% c("chrX","chrY")]) table(keep) mSetSqFlt <- mSetSqFlt[keep,]过滤掉所有SNP相关的探针,作者建议用
minfi包的dropLociWithSnps函数,除了去除默认的SNPs位点外,还可以去除那些minor allele frequencies高于阈值的位点;注:该函数是默认去除SNPs出现在CpG or single base extension (SBE) site的探针mSetSqFlt <- dropLociWithSnps(mSetSqFlt, snps = c("CpG", "SBE")) mSetSqFlt去除cross-reactive的探针,也就是multi-hit探针,即映射到多个位置的;这探针list是Chen et al.(2013)发表的,所以只要出现在这list上面的探针都可以去除了
dataDirectory <- "E:/R-3.4.1/library/methylationArrayAnalysis/extdata/" xReactiveProbes <- read.csv(file=paste(dataDirectory, "48639-non-specific-probes-Illumina450k.csv", sep="/"), stringsAsFactors=FALSE) keep <- !(featureNames(mSetSqFlt) %in% xReactiveProbes$TargetID) mSetSqFlt <- mSetSqFlt[keep,] mSetSqFlt
做完上述过滤和标准化后,作者建议再看下MDS plot,看看样本之间的联系是否发生了改变。对这个测试数据而言,第一个主成分上样本不像之前那么集中了,作者怀疑是由于去除了SNP关联的探针的缘故
作者使用minfi包的getM和getBeta来分别计算M-values和Beta-values,作者认为M-values具有更好的统计特性,更适合用于进行下游的统计分析(差异分析等);而Beta-values更加容易解释,更能说明生物学上的意义
mVals <- getM(mSetSqFlt)
bVals <- getBeta(mSetSqFlt)并对两者分别做了密度分布图
par(mfrow=c(1,2))
densityPlot(bVals, sampGroups=targets$Sample_Group, main="Beta values", legend=FALSE, xlab="Beta values")
legend("top", legend = levels(factor(targets$Sample_Group)), text.col=brewer.pal(8,"Dark2"))
densityPlot(mVals, sampGroups=targets$Sample_Group, main="M-values", legend=FALSE, xlab="M values")
legend("topleft", legend = levels(factor(targets$Sample_Group)), text.col=brewer.pal(8,"Dark2"))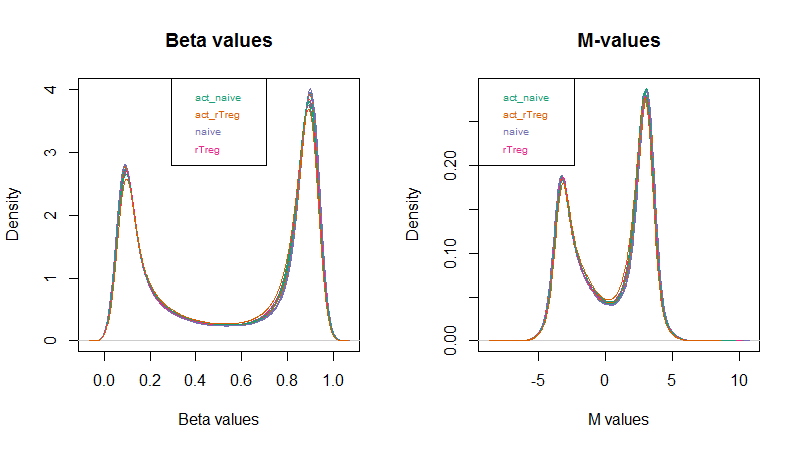
差异甲基化分析
作者选了M-values作为后续分析的矩阵,对于差异甲基化位点的分析,作者没有用minfi包的dmpFinder函数,而是用了limma包的linear model来分析。所以按照limma的分析思路:
构建分组矩阵
cellType <- factor(targets$Sample_Group) individual <- factor(targets$Sample_Source) design <- model.matrix(~0+cellType+individual, data=targets) colnames(design) <- c(levels(cellType),levels(individual)[-1])构建两两比较矩阵
contMatrix <- makeContrasts(naive-rTreg, naive-act_naive, rTreg-act_rTreg, act_naive-act_rTreg, levels=design)差异分析
fit <- lmFit(mVals, design) fit2 <- contrasts.fit(fit, contMatrix) fit2 <- eBayes(fit2)将位点信息和
limma差异分析的结果合并在一起,这里以naive - rTreg为例ann450kSub <- ann450k[match(rownames(mVals),ann450k$Name), c(1:4,12:19,24:ncol(ann450k))] DMPs <- topTable(fit2, num=Inf, coef=1, genelist=ann450kSub) head(DMPs)
除了差异甲基化位点分析,作者还提了怎么进行DMR(Differential Methylation Regions)分析;作者总结了几个可用来DMR分析的bioconductor包,如:minfi包的bumphunter函数,DMRcate包的dmrcate函数。由于作者觉得bumphunter函数运算速度过慢,除非使用多线程并行运算(如doParallel包),所以选择用dmrcate函数;并且由于这个函数也是基于limma,所以可以直接使用上面的design和contMatrix
先用
DMRcate包的cpg.annotate函数做单个甲基化位点的差异分析以及注释myAnnotation <- cpg.annotate(object = mVals, datatype = "array", what = "M", analysis.type = "differential", design = design, contrasts = TRUE, cont.matrix = contMatrix, coef = "naive - rTreg", arraytype = "450K")接着用
dmrcate函数在上面单个甲基化位点分析的基础上进行合并,从而识别出DMR(Differential Methylation Regions)DMRs <- dmrcate(myAnnotation, lambda=1000, C=2) head(DMRs$results)用
DMR.plot函数进行可视化,看看这些DMRs在基因组上的展现形式# convert the regions to annotated genomic ranges data(dmrcatedata) results.ranges <- extractRanges(DMRs, genome = "hg19") # set up the grouping variables and colours groups <- pal[1:length(unique(targets$Sample_Group))] names(groups) <- levels(factor(targets$Sample_Group)) cols <- groups[as.character(factor(targets$Sample_Group))] samps <- 1:nrow(targets) # draw the plot for the top DMR par(mfrow=c(1,1)) DMR.plot(ranges=results.ranges, dmr=1, CpGs=bVals, phen.col=cols, what = "Beta", arraytype = "450K", pch=16, toscale=TRUE, plotmedians=TRUE, genome="hg19", samps=samps)
Summary
我这只介绍了这篇Bioconductor workflow for analysing methylation array的部分内容,后续还有讲如何用Gviz包定制化展示甲基化数据,如何用missMethyl包做后续的GO/KEGG分析,还以一个肿瘤甲基化研究的文章为例,展示如何用甲基化来分析肿瘤样本的整个流程(整体上与上述的测试例子类似,但也有点不同)。总之,是一篇值得从头到尾好好看的DNA甲基化分析流程的文章!
注:上述文字和代码均来自bioconductor的文章,我只以自己的理解再次记录下,可能有理解不到的地方,所以还是推荐看原文!
本文已在《生信技能树》公众号上投稿,并授权转载
本文出自于http://www.bioinfo-scrounger.com转载请注明出处