我们常见的转录组表达分析一般都是将reads比对至参考基因组或者转录组上,然后在基因或者转录本水平上定量表达丰度。
如果是在基因水平上的定量分析,那么一般可以选择用HISAT2比对,HTSeq-count定量;如果是在转录本水平,可以先用HISAT2比对,然后Stringtie组装转录本(假如只对已知isoforms定量,那么这步一般是可以省略的),再用RSEM或者eXpress定量表达丰度。
之前一般我也是这么做的,其实以上步骤在转录本定量中属于同一类:Alignment-based transcript quantification,还有一类则是:Alignment-free transcript quantification;后者相比前者而言,省略了比对这一步骤(有些软件是只进行'轻度'比对),从而直接将reads分配到转录本上;后者相比前者spliced alignment能极大的减少计算机资源的使用,代表软件有Sailfish,Salmon,kallisto,而这次我则尝试下Salmon
测试数据准备
之前的使用的测试数据太老了,这次就下载个新的RNA-Seq数据,双端测序100bp,Illumina HiSeq 2500,地址如下:https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP124620
然后用sratoolkit软件将sra格式转化为fastq格式,接着用fastqc做下质控,发现有TruSeq Adapter,因而选trimmomatic去接头和低质量碱基
for i in $(seq 49 52);do java -jar ~/biosoft/trimmomatic/Trimmomatic-0.36/trimmomatic-0.36.jar PE -phred33 ./SRR62690${i}_1.fastq ./SRR62690${i}_2.fastq SRR62690${i}.clean_1.fastq SRR62690${i}.unpaired_1.fastq SRR62690${i}.clean_2.fastq SRR62690${i}.unpaired_2.fastq ILLUMINACLIP:/home/anlan/biosoft/trimmomatic/Trimmomatic-0.36/adapters/TruSeq3-PE.fa:2:30:10 LEADING:20 TRAILING:20 SLIDINGWINDOW:4:20 MINLEN:36 -threads 5;done软件下载及安装
Salmon最新版下载地址:https://github.com/COMBINE-lab/salmon/releases
安装就更加简单了,因为作者提供了二进制版本,解压缩后,即可使用bin文件夹下的执行程序salmon
tar zxvf Salmon-0.9.1_linux_x86_64.tar.gz
cd Salmon-latest_linux_x86_64/bin也可以用bioconda进行安装,而且也是最新版(v0.9.1)
conda install -c bioconda salmon软件使用
使用前先参观下Salmon的Github上的文档https://combine-lab.github.io/salmon/,一条鲑鱼?三文鱼?反正是一条鱼展示在页面顶部。。
按照其说法,Salmon使用了新的算法能更快更精确的估计RNA-SEQ数据的表达量,并且只需要很少的内存。并且Salmon使用了expressive and realistic模型以及结合experimental attributes and biases使得其推测的表达量更加符合实际的RNA-SEQ数据
在使用Salmon前我们需要对其有一定的了解
现在Salmon支持两种模式将reads mapping到转录本上:第一种是
quasi-mapping-based mode,其是一种最新的以及快速准确的定量转录本的方法,不需要像传统的那样需要通过比对才能将reads mapping到转录本上;第二种则是alignment-based mode,跟类似RSEM软件一样,提供比对后的bam/sam文件即可对转录本进行定量第一种方法需要对转录本建index,第二种方法则不需要
quasi-mapping模式是在RapMap tools中实现的,其可用于transcript quantification,clustering of de novo assembled transcripts and filtering of potential target transcriptsRapMap: a rapid, sensitive and accurate tool for mapping RNA-seq reads to transcriptomes
下面主要是quasi-mapping模式的使用
首先建索引
~/biosoft/salmon/Salmon-latest_linux_x86_64/bin/salmon index -t ~/reference/transcriptome/hg38/Homo_sapiens.GRCh38.cdna.all.fa -i hg38_transcript --type quasi -k 31-k是指k-mers长度,默认是31,the k size selected here will act as the minimum acceptable length for a valid match,减少其值能略微增加mapping的灵敏度;按照作者的说法,如果你的reads大于75bp,那么k设置为31是较好的选择,如果reads低于75可略微减少K值(值得注意的是,这里的K值跟alignment-based mode中的K值是不一样的)
转录本定量
for i in $(seq 49 52);do ~/biosoft/salmon/Salmon-latest_linux_x86_64/bin/salmon quant -i ~/reference/index/salmon/hg38_transcript/ -p 6 --libType IU -1 SRR62690${i}.clean_1.fastq -2 SRR62690${i}.clean_2.fastq -o SRR62690${i}.quant;done重要参数
--libType是一个比较重要的参数,跟文库类型相关;如果
--libType A则说明让Salmon自动来检测RNA-SEQ文库类型当然我们也可以手动设置,比如第一部分是说明文库的reads的方向(The first part of the library string (relative orientation))
I = inward O = outward M = matching第二部分是说明文库是否是特异性链文库
S = stranded U = unstranded如果是特异性链的话,还有继续设置read1来自哪一条链
F = read 1 (or single-end read) comes from the forward strand R = read 1 (or single-end read) comes from the reverse strand综上所述,如果RNA-SEQ是mRNA双端测序普通建库,那么选择的就是IU (an unstranded paired-end library where the reads face each other)
如果不是太懂的话,看官网文档的解释或者下面这张图,一般能懂了
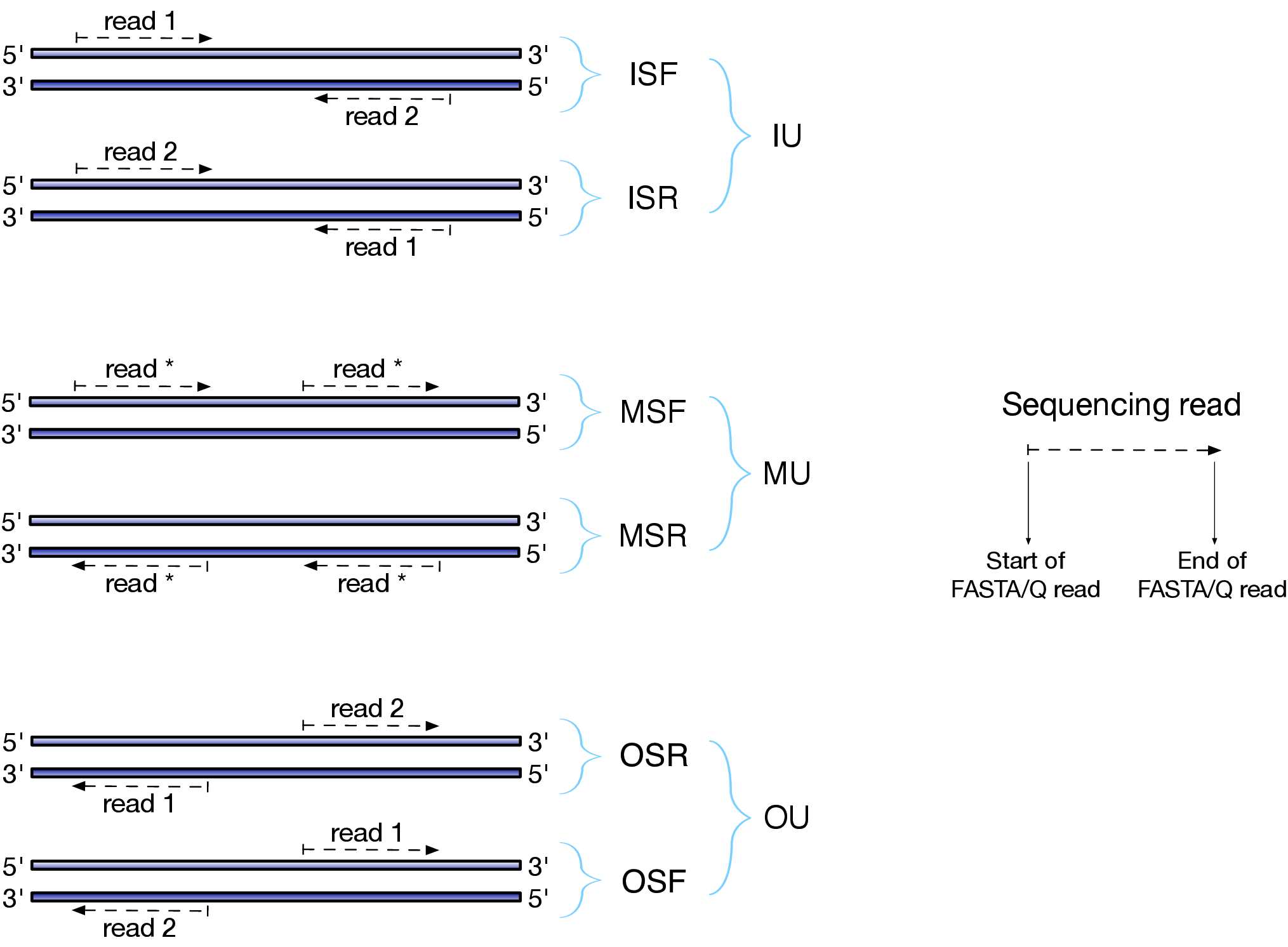
Salmon_libType 除了
--libType参数外,还有一些参数也是蛮有意思的,值得解读下:--incompatPrior用于设定incompatible mappings是否被舍去;这里的incompatible mappings是指与设置的libType不一样的比对;如果是默认值(非常小的值但不等于0)表示如果一个incompatible mapping对一个fragment来说是唯一的一个mapping(有点绕了。。),那么这个fragment算是mapping上了;如果设置为0则表示incompatible mapping全部被舍弃(类似RSEM);如果设置为1则表示不做上述的处理,即incompatible mapping不被舍弃--useVBOpt用于设定将默认使用的标准EM algorithm变为使用variational Bayesian EM algorithm;前者倾向于sparser estimates(即多数转录本估计值为0丰度);后者倾向于多数转录本估计值为非零丰度--writeMappings将mapping结果以SAM格式输出,默认是stdout输出到屏幕;可以通过这个SAM文件来查看下到底有哪些reads比对上了,是以什么方式(Flags)mapping上的grep -v '^@' xxx.sam |cut -f 2|sort |uniq -c |sort -rn |less根据统计的FLAGS,可以看到大部分reads的alignment信息都是Not primary alignment,mapped reads也有16479800和16381524
52541802 419 52541802 339 52122426 403 52122426 355 16479800 83 16479800 163 16381524 99 16381524 147 281210 437 281210 377 275740 441 275740 373 255515 405结果文件
所有结果文件都输出在一个文件夹中(以-o参数设定),其中最主要的文件要属
quant.sf文件,其记录了每个转录本的count数以及对应的TPM值,其他几列信息根据列名就能轻松看懂的;其中最主要的还是NumReads列: >It is an “estimate” insofar as it is the expected number of reads that have originated from each transcript given the structure of the uniquely mapping and multi-mapping reads and the relative abundance estimates for each transcript
顺便统计下总count数
perl -alne '$sum += $F[4]; END {print $sum}' quant.sf从上述结果以及结合FLAGS结果能看出,Salmon对于那些multimpping reads还是考虑进去的,但是count计数时是考虑其中的primary alignment?即只选multimapping reads中most likely的reads,这就考验算法的时候了(Salmon默认是用expectation maximization(EM)的)
meta_info.json文件也是值得一看的文件,里面记录一些信息,如num_processed(检测到的参与计算的reads),num_mapped(mapping上的reads数,其实就是总count数)以及如果--libType设置A的话,可以看library_types来确定软件检测到的library type是哪种ambig_info.tsv文件可以看每个转录本对应的Unique and ambiguous count
Summary
Salmon还支持alignment-based mode,但是作者建议是将reads align到转录本上(类似RSEM和eXpress),而不是align到基因组上(使用Hisat2等),具体使用可以看官网说明
最后再推荐个转录本定量的PDF,可以看看,Accurate and Fast Transcript (and gene) Quantification
参考资料:
http://salmon.readthedocs.io/en/latest/salmon.html#
本文出自于http://www.bioinfo-scrounger.com转载请注明出处