去年的这个时候,我参加了生信技能树Jimmy办的线上R语言处理表达芯片教程,这也是我进入生信领域,第一个学习的完整流程。那时对生信的一些基础概念还是处于模糊的状态,只是略懂了点R语言,勉强的听完了整个教程。 后来似乎也再也没接触过芯片数据了,对于芯片数据的概念还停留在表达矩阵那块,以致有次在找公共数据时没找到表达矩阵,只找到了芯片的raw data,却因无法分析而无能为力(现在看来是由于在学习教程时并没有完全吃透)
有幸不久之前看到一篇推荐的芯片教程Differential gene expression analysis I (microarray data),看了后感觉受益不少,然后再从头看了一遍Jimmy的教程,粗略了解Affymetrix平台芯片该如何进行前期预处理以及获取表达矩阵
基础知识
一组探针(或探针组probe set)来自于一个基因,通常是由11对探针组成,每对探针是由匹配探针(PM)和错配探针(MM)组成,叫做探针对(Probe Pair),样本RNA则叫做靶标。芯片中MM探针作用是检测非特异杂交信号,理论上,MM只有非特异杂交信号,而不会有特异性杂交。MM的信号值永远小于其对应的PM信号值,因此可以做一个PM-MM或PM/MM即可去除背景噪声的影响
数据下载
以芯片教程中的GSE27114作为实例进行后续操作。在GSE27114里有两个study,一个是比较旧的Affymetrix平台数据(GPL1261):Affymetrix Mouse Genome 430 2.0 Array(Affymetrix 3’ end arrays),另一个是相对较新的平台数据(GPL6246):Affymetrix Mouse Gene 1.0 ST Array(Affymetrix arrays Whole Transcript Gene Arrays),下面先以GPL1261为例:
在数据下载之前还需要先了解自己需要哪一个芯片数据,也就是说是需要芯片原始数据(CEL文件)还是表达矩阵数据(matrix)
如果是只想获得芯片数据已经做完标准化等预处理过的表达矩阵,那么可以使用GEOquery包的getGEO函数,表达矩阵文件的压缩包也会下载到R的工作目录下
library(GEOquery)
gset <- getGEO("GSE27114", destdir = ".")由于GSE27114里有2个study,所以我先看看有哪两个study
names(gset)
##[1] "GSE27114-GPL1261_series_matrix.txt.gz" "GSE27114-GPL6246_series_matrix.txt.gz"由于下面会以GPL1261为例,所以我先取出第一个study,可以看到gset_one的数据是储存在ExpressionSet对象中,这个对象里包含了N多的信息,可以通过slotNames列出可以查看的信息,然后根据列出来的函数一一查看,比如assayData用于保存表达数据信息,experimentData用于保存实验设计信息
gset_one <- gset[[1]]
class(gset_one)
##[1] "ExpressionSet"
##attr(,"package")
##[1] "Biobase"接下来就可以通过exprs函数获取表达矩阵,有45101个探针,6个样本
exprSet <- exprs(gset_one)
dim(exprSet)
##[1] 45101 6还可以通过pData获取这个study的更多详细信息,比如样本信息、分组信息、rawdata信息,比如这个例子就有32列信息。。。
pdata <- pData(gset_one)
dim(pdata)
##[1] 6 32当然如果只想获取表达矩阵,并且其他信息并不想知晓(或者已经在GEO网站上查到),那么通过GEO网站手动下载表达矩阵也是可以的:点击GSE27114网页下方的Series Matrix File(s)即可进入ftp下载界面ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE27nnn/GSE27114/matrix/,而且这个ftp的URL可以根据你的GSE号自行进行修改,再配合脚本就可以批量下载N个GSE样本的表达矩阵,很好用。
如果是想获取GSE27114的RAW DATA,那么可以选择使用oligo包(Affymetrix 平台),主要用于读取Affymetrix平台的基因表达数据,处理为表达矩阵;如果是Illumina平台,教程则推荐使用beadarray或lumi
首先通过download.file函数下载raw data,至于下载URL如何获取有两个途径:
通过上述的pdata里查看,有一列是各个样本的rawdata下载地址,然后写个循环即可批量下载数据
pdata$supplementary_file通过GSE27114网站的最下方GSE27114_RAW.tar的,点击http即可下载所有文件的压缩包(网速慢或者不稳定可以首选这个,至少下载一个算一个,慢慢来。。。)
数据读入及标准化
将所有样本的raw data文件放置在R工作目录下(Microarray文件夹),然后用list.celfiles函数查看所有文件,再用read.celfiles函数读入R中,并将上述的pdata信息也一并写入
library(oligo)
file_CELs <- list.celfiles("Microarray", listGzipped = TRUE, full.name = TRUE)
rawAffy <- read.celfiles(filenames = file_CELs, phenoData = phenoData(gset_one), sampleNames = rownames(pData(gset_one)))注:读入文件时会检查R中是否安装了该数据对应平台的注释包,比如这个例子需要这个pd.mouse430.2包
粗略看下,rawAffy数据中包含了芯片上所有探针的强度值信息
head(exprs(rawAffy), n = 2)我们可以拿这个数据来进行质控,但是一般质控会在芯片公司那边就做了,所以我们拿公共数据的用的话,一般也不用进行质控了。所以我们接下来就对rawAffy数据进行标准化处理,因为每个样本芯片的绝对光密度是不一样的,不做处理的话无法将各个样本间进行数据分析,同时也是为了消除测量间的非实验差异
芯片常用的标准化方法也有好几种(如:MAS5),这里就不一一尝试了,就拿最常用的RMA(Robust Multichip Average algorithm),引用芯片教程,RMA标准化过程主要分为3步:
- Background correction (removes array auto-fluorescence ) 背景矫正
- Quantile normalization (makes all intensity distributions identical) 标准化
- Probeset summarization (calculates one representative value per probeset) 汇总
由于rawAffy是ExpressionFeatureSet对象,则rma函数默认是probeset level
class(rawAffy)
##[1] "ExpressionFeatureSet"
##attr(,"package")
##[1] "oligoClasses"
eset <- rma(rawAffy)我们可以看看标准化后的结果
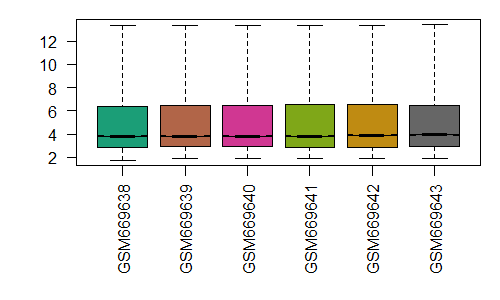
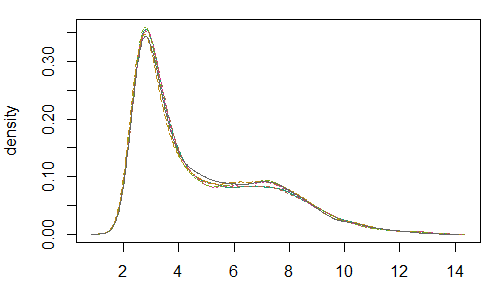
最后我们需要得到表达矩阵,也是用exprs函数,从结果上来看,跟之前getGEO得到的表达矩阵是一样的(说明用的是同一种标准化方法?以及相同的QC处理?)
exprSet <- exprs(eset)
dim(exprSet)
##[1] 45101 6参考资料:
Differential gene expression analysis I (microarray data)
本文出自于http://www.bioinfo-scrounger.com转载请注明出处