做完前面两部分实战总结ChIP-Seq分析小实战(一) ChIP-Seq分析小实战(二),这个实战教程也剩下了最后的peak注释以及可视化了 简单的说,这次的chip-seq实战可以分为以下几个步骤:
- 测序数据的下载及质控
- mapping reads(这次使用的是bowtie2)
- peak calling(这次使用的是MACS2)
- peak注释及可视化(ChIPseeker包、deeptools)
peak 注释
peak注释个人简单的理解为寻找chip-seq所对应的靶基因,先看下文章如何定义靶基因的:
Genes with a peak within the gene body, or within 2.5 kb from the TSS, were considered to be target genes
也就是说在基因区间内或距离TSS 2.5kb的peak的区域被认为是靶基因
我们先看文章中的Figure 1A:
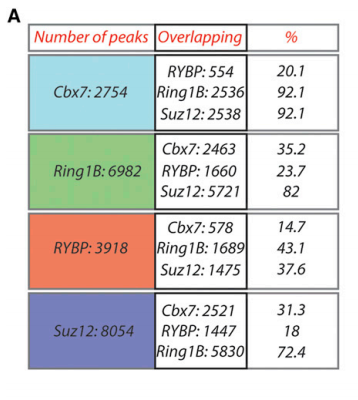
其表示Cbx7,Ring1B,RYBP和Suz12之间peaks的overlapping的情况,那么我们就按照文章中所说的,用bedtools看看BYBP对其他3个样本peaks的重叠情况 >ChIP-seq profiles around the TSSs were generated for each IP by calculating the average coverage in each position normalized for the total number of mapped reads with the BED tools package
由于自己之前跑出来的peaks文件的在软件参数以及参考基因选择上跟文献会有不小的差别,因此下面统一用GSE网站上下载下来的peaks文件做后续分析
wc -l *peaks.bed
2754 Cbx7_peaks.bed
6982 Ring1b_peaks.bed
6872 RYBP_peaks.bed
8054 Suz12_peaks.bed然后用bedtools对RYBP和Cbx7样本的peaks取overlap(才发现RYBP_peaks.bed的peaks数目跟文献的竟然不一样。。。),所以取Cbx7_peaks.bed和Ring1b_peaks.bed为例:
bedtools intersect -a Ring1b_peaks.bed -b Cbx7_peaks.bed >Ring1b_cbx7_coverage.bed结果为2647个overlapping,跟文献的2463还是有点差距的,按照我现在的知识只能理解为工具上的使用的差异了。接着按照文献所示的图Figure 1B,找两个peaks文件的靶基因的交集(即venn图)

这里准备使用ChIPseeker包来根据peaks对靶基因进行注释。PS.这里我将使用自己MACS2跑出来的xxx_peaks.narrowPeak;不使用作者上传的是peaks文件是因为ChIPseeker包不识别。。。并且还需要将xxx_peaks.narrowPeak文件改为bed后缀,不然ChIPseeker包读进去会将第一行作为行名(我暂时还没在包说明文档中找到为啥会出现这种情况),所以我3个文件为下述: >cbx7_peaks.narrowPeak.bed
>ring1B_peaks.narrowPeak.bed
>suz12_peaks.narrowPeak.bed
首先读入必须的包和上述3个文件
library(ChIPseeker)
library(org.Mm.eg.db)
library(TxDb.Mmusculus.UCSC.mm10.knownGene)
library(VennDiagram)
cbx7 <- readPeakFile("cbx7_peaks.narrowPeak.bed")
ring1B <- readPeakFile("ring1B_peaks.narrowPeak.bed")
suz12 <- readPeakFile("suz12_peaks.narrowPeak.bed") 接着我们需要用peakAnno函数对其进行peak注释,这里的注释主要可以分为三种,genomic注释、nearest gene注释以及peak上下游注释,具体解释可参照包作者写的文档peak注释。按照文章中对靶基因的要求,我们选择前两者注释即可
cbx7_peakAnno <- annotatePeak(cbx7, tssRegion = c(-2500, 2500), TxDb = TxDb.Mmusculus.UCSC.mm10.knownGene, annoDb="org.Mm.eg.db")
ring1B_peakAnno <- annotatePeak(ring1B, tssRegion = c(-2500, 2500), TxDb = TxDb.Mmusculus.UCSC.mm10.knownGene, annoDb="org.Mm.eg.db")
suz12_peakAnno <- annotatePeak(suz12, tssRegion = c(-2500, 2500), TxDb = TxDb.Mmusculus.UCSC.mm10.knownGene, annoDb="org.Mm.eg.db") 然后提取出cbx7和ring1B的注释到的gene,绘制venn图
venn_list <- list(cbx7=as.data.frame(cbx7_peakAnno)$geneId,
ring1B=as.data.frame(ring1B_peakAnno)$geneId)
venn.diagram(venn_list, imagetype = "png", fill = c("blue", "green"),
alpha = c(0.5, 0.5) ,filename = "venn.png")
虽然结果和文章的有较大的差距,但是考虑到软件参数以及参考基因组的不同,也就算了。。
peak 注释可视化
Peak注释可视化即对注释到的peaks进行可视化展示,除了上面的靶基因的venn图,文章还对ChIP-seq profiles进行了展示 >An area of 5 kb surrounding each TSS that was associated to one or more ChIP-seq (RYBP, Ring1B, Cbx7, Suz12, or H2AK119ub) was used to calculate the ChIP-seq profile and the whole ChIP-seq coverage.
使用ChIPseeker包可视化
可以用
covplot看一下ChIP-seq peaks在染色体上的分布图,比如cbx7的covplot(cbx7, weightCol="V5")做cbx7、ring1B和suz12样本共有的靶基因在TSS附近的profile图(代码续上述的靶基因注释)
#先获得共有靶基因的 common_gene <- Reduce(intersect, list(cbx7=as.data.frame(cbx7_peakAnno)$geneId, ring1B=as.data.frame(ring1B_peakAnno)$geneId, suz12=as.data.frame(suz12_peakAnno)$geneId)) #然后提取出每个样本的在共有靶基因上的peaks,从而得到tagMatrix并绘图 cbx7_df <- as.data.frame(cbx7_peakAnno) cbx7_peak <- GRanges(cbx7_df[cbx7_df$geneId %in% common_gene, 1:12]) ring1B_df <- as.data.frame(ring1B_peakAnno) ring1B_peak <- GRanges(ring1B_df[ring1B_df$geneId %in% common_gene, 1:12]) suz12_df <- as.data.frame(suz12_peakAnno) suz12_peak <- GRanges(suz12_df[suz12_df$geneId %in% common_gene, 1:12]) list_peak <- list(cbx7=cbx7_peak, ring1B=ring1B_peak, suz12=suz12_peak) promoter <- promoters(TxDb.Mmusculus.UCSC.mm10.knownGene, upstream = 2500, downstream = 2500) tagMatrix <- lapply(list_peak, getTagMatrix, window=promoter) plotAvgProf(tagMatrix, xlim=c(-2500, 2499), xlab="Genomic Region (5'->3')", ylab = "Read Count Frequency")上述的代码有点冗余,但是凑合用着了;图如下所示,也跟文献的更加不像了。。。至于为啥用2499,是因为用2500报错,应该是跟我输入文件有关。。
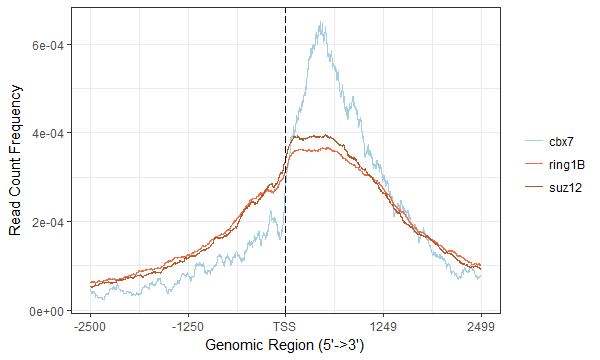
ChIP-seq_profile.png 除了profile图,还可以画画热图,图就不展示了
tagHeatmap(tagMatrix, xlim=c(-2500, 2499), color=NULL)使用deeptools可视化
deeptools也可以一个可以对ChIP-seq结果进行可视化展示的软件,展现形式也差不多,也有profile图和热图等,下面粗略用一下
以cbx7样本为例,先用
bamCoverage将bam文件转化为bw文件bamCoverage -p 4 -b cbx7.bam -o cbx7.bw然后用
computeMatrix将peaks定位到mm10.bed文件上computeMatrix reference-point --referencePoint TSS -b 2500 -a 2500 -S cbx7.bw -R ~/annotation/mm10/mm10.bed --skipZeros -o cbx7_TSS.gz --outFileSortedRegions cbx7_region_genes.bed最后绘制全部peaks在TSS附件的热图和profile图
plotHeatmap -m cbx7_TSS.gz -out cbx7.png plotProfile --dpi 720 -m cbx7_TSS.gz -out cbx7_profile.pdf --plotFileFormat pdf如果是多个样本,可下述用
computeMatrix一起读入.bw文件,再绘制图片computeMatrix reference-point --referencePoint TSS -b 2500 -a 2500 -S ./*.bw -R ~/annotation/mm10/mm10.bed --skipZeros -o merged_TSS.gz --outFileSortedRegions merged_region_genes.bed plotHeatmap -m merged_TSS.gz -out merged.png plotProfile --dpi 720 -m merged_TSS.gz -out merged_profile.pdf --plotFileFormat pdf
Summary
这次实战只是对ChIP-seq的一般流程进行初步的了解,还了解了ChIP-seq的一些基本概念。虽然从文献角度来说,这篇文献进行了些其他的研究,但是这里就不在对其重现了(一方面是作者上传数据的限制,另一方面则的能力有限~)。再者,这文献研究的是PRC1这个蛋白复合物,而ChIP-Seq似乎在组蛋白修饰和转录因子方面的研究应用更多点,可能还需进行找篇文献对这两方面进行尝试,加深下对ChIP-seq方法的理解
参考资料:
http://bioconductor.org/packages/devel/bioc/vignettes/ChIPseeker/inst/doc/ChIPseeker.html
http://deeptools.readthedocs.io/en/latest/content/list_of_tools.html
http://www.jianshu.com/p/a7b6ce208f98
本文出自于http://www.bioinfo-scrounger.com转载请注明出处