RSeQC是发表于2012年的一个RNA-Seq质控工具,属于python包。
可用于评估RNA-Seq实验的各个方面,比如sequence quality, GC bias, polymerase chain reaction bias, nucleotide composition bias, sequencing depth, strand specificity, coverage uniformity and read distribution over the genome structure 主要输入文件为RNA-Seq mapping后的SAM or Bam文件
安装
方法1:
git clone --branch fresh https://github.com/MonashBioinformaticsPlatform/RSeQC.git
cd RSeQC
python setup.py install
pip install numpy方法2:(我一般使用bioconda来安装python包的软件,因为不会报错!!!)
conda install -c rseqc使用
bam_stat.py用于查看比对片段的统计结果,拿我转录组实战的一个bam文件作为例子:bam_stat.py -i SRR3589956.bam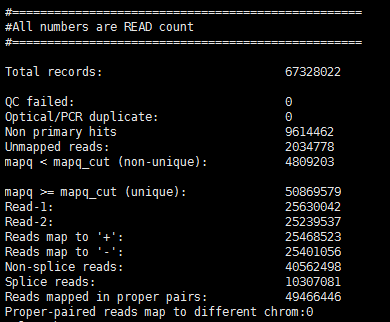
RSeQC_bam_stat bam2fq.py顾名思义就是将bam文件转化为fastq格式。。。话说只要懂bam格式的人,一般也会自己写脚本转化,当然有RSeQC的话,能省点时间bam2wig.py用于将bam转化为wig/bigWig格式,但是这个格式本来就是UCSC定义的,因此UCSC也有转化工具,注意的是:使用这个命令前需要将bam文件进行排序然后建索引samtools -@ 5 SRR3589956.bam SRR3589956.sorted.bam samtools index SRR3589956.sorted.bam bam2wig.py -s hg19.chrom.sizes -i SRR3589956.sorted.bam -o out -uhg19.chrom.sizes是一个tab分割的文件,总共有两列,第一列是染色体名称(chr1),第二列是染色体对应的size
divide_bam.py字面上也很好理解,就是分割bam文件(总共有m个比对结果),比如你想分割成5个小的bam文件,那么每个小bam文件就有m/5个比对结果divide_bam.py -n 5 -i SRR3589956.bam -o outputgeneBody_coverage.py这个比较常用了,一般用于展示reads在基因组(转录本)上的覆盖情况,但是需要对应基因组的bed文件,一般可以从标准的gtf文件转化而来,即可用gtf2bed软件,也可以用shell(网上找的),这里就尝试下:##gtf转化为bed cat reference/genome/hg19/gencode.v26lift37.annotation.gtf |awk 'OFS="\t" {if($3=="transcript") {print $1,$4-1,$5-1,$12,$6,$7}}' |tr -d '";' >hg19.bed然后发现必须要12列的bed文件才能用,简单版的不行。。。所以只好用ucsc的gtfToGenePred配合shell来将gtf转化为bed12.。。
gtfToGenePred -genePredExt -geneNameAsName2 ~/reference/genome/hg19/gencode.v26lift37.annotation.gtf gene.tmp awk '{print $2"\t"$4"\t"$5"\t"$1"\t0\t"$3"\t"$6"\t"$7"\t0\t"$8"\t"$9"\t"$10}' gene.tmp > hg19.bed12最后,运行geneBody_coverage.py
geneBody_coverage.py -r hg19.bed12 -i SRR3589956.bam -o outputread_quality.py这个用于查看bam/sam文件中reads的测序质量,展示的图片跟fastq那种差不多read_quality.py -i SRR3589956.bam -o outputread_distribution.py这个我觉得比较好使,可以查看统计read分布在基因组上一些的区域的的数目,比如CDS exon, 5’UTR exon, 3’ UTR exon, Intron, Intergenic regions这些区域read_distribution.py -i SRR3589956.bam -r hg19.bed12
剩下的都是一些我暂时感觉用到的命令了,或者跟其他一些软件功能重复了
详细的可以参照http://pythonhosted.org/RSeQC/#usage-information 有非常详细的介绍以及结果展示
参考文章:
https://github.com/Czh3/NGSTools/blob/master/script/gtf2bed12.sh
http://www.agctu.org/question/160
本文出自于http://www.bioinfo-scrounger.com转载请注明出处